

Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders
- PMID: 34239129
- PMCID: PMC8521603
- DOI: 10.1038/s41593-021-00886-6
Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders
Abstract
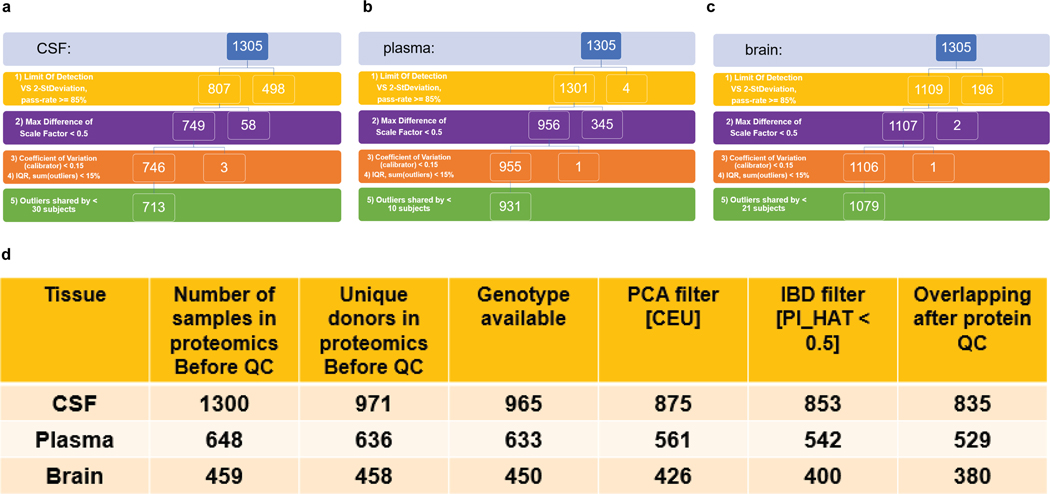
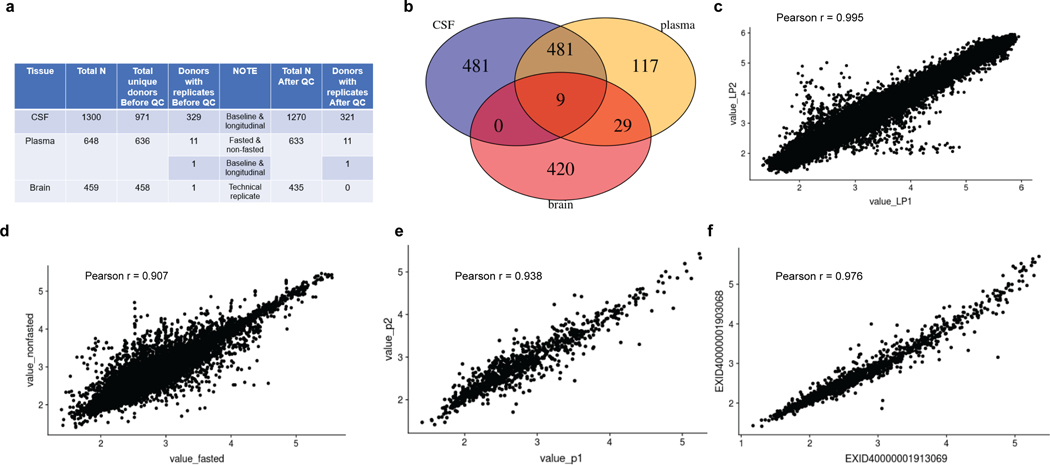
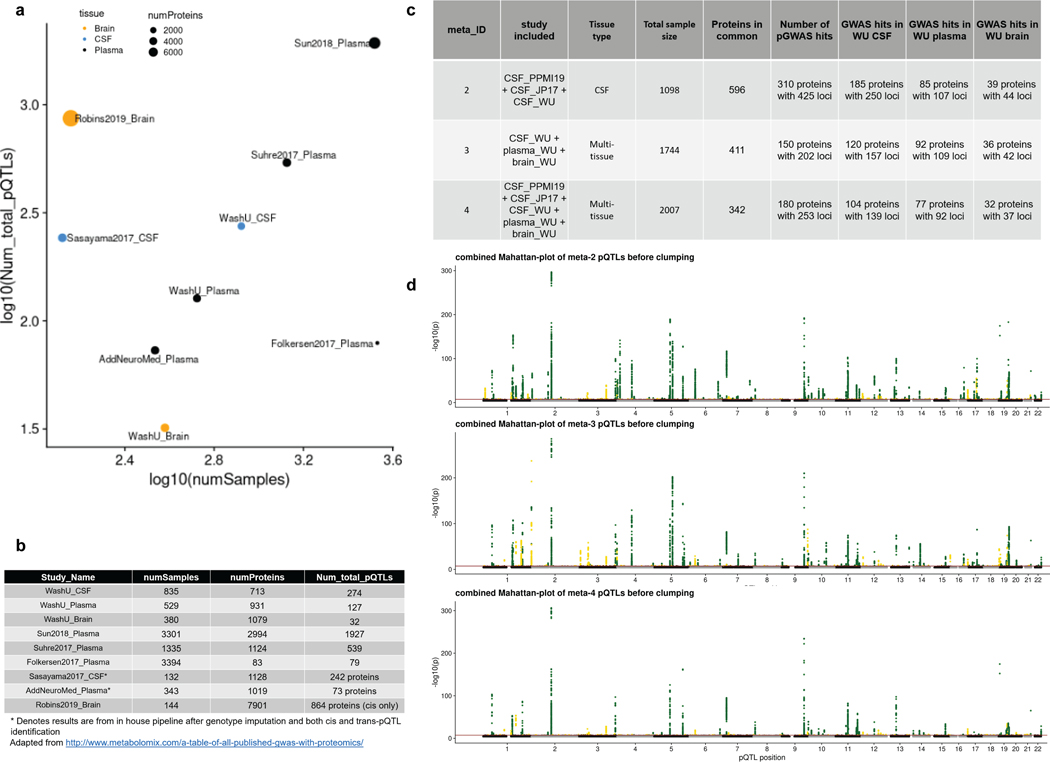
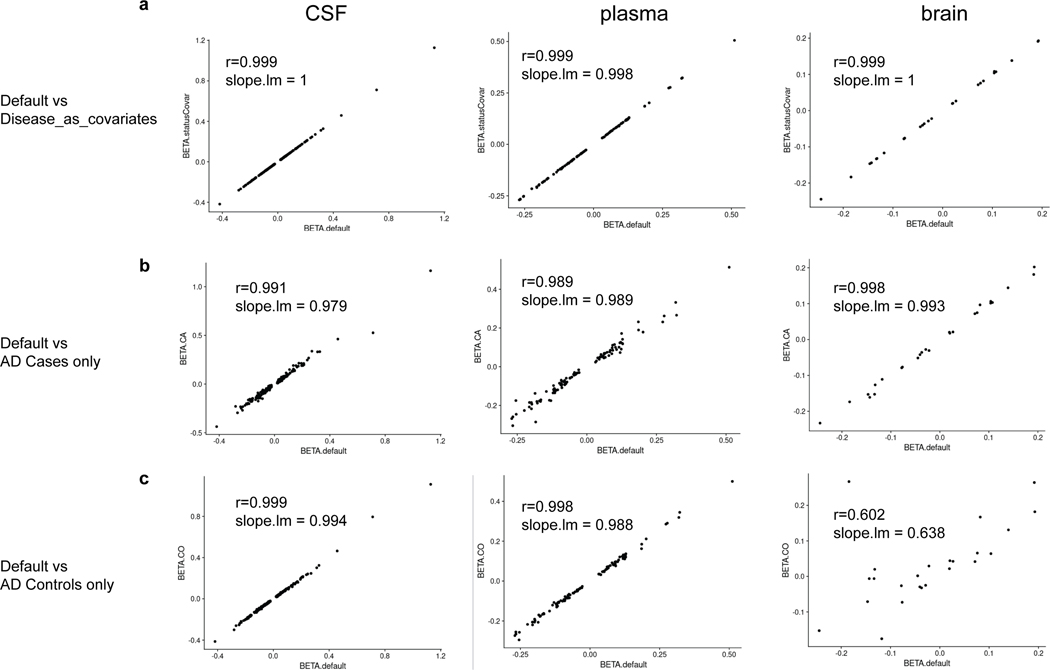
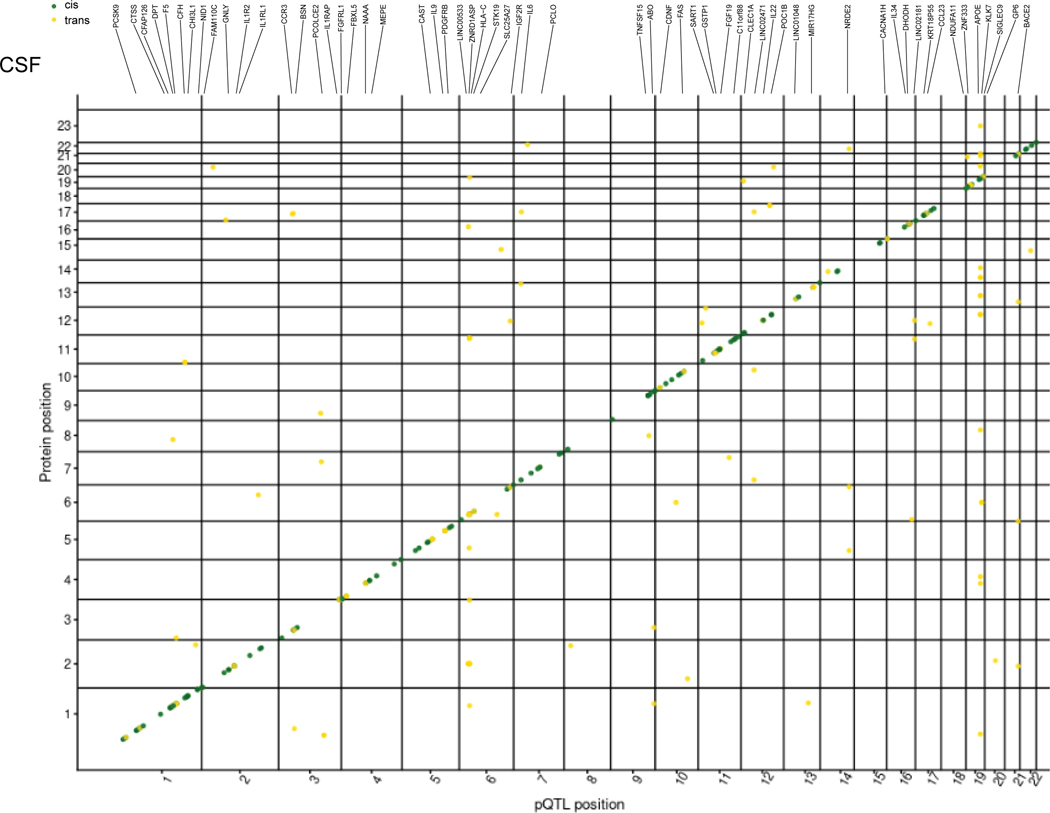
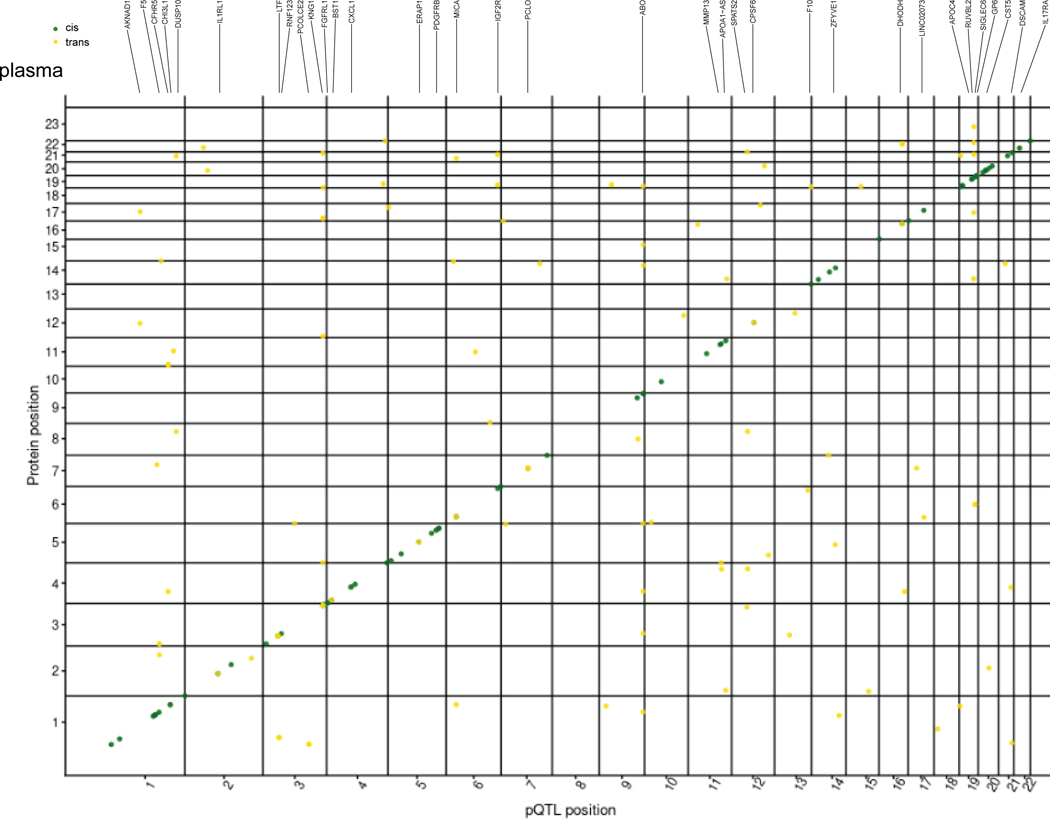
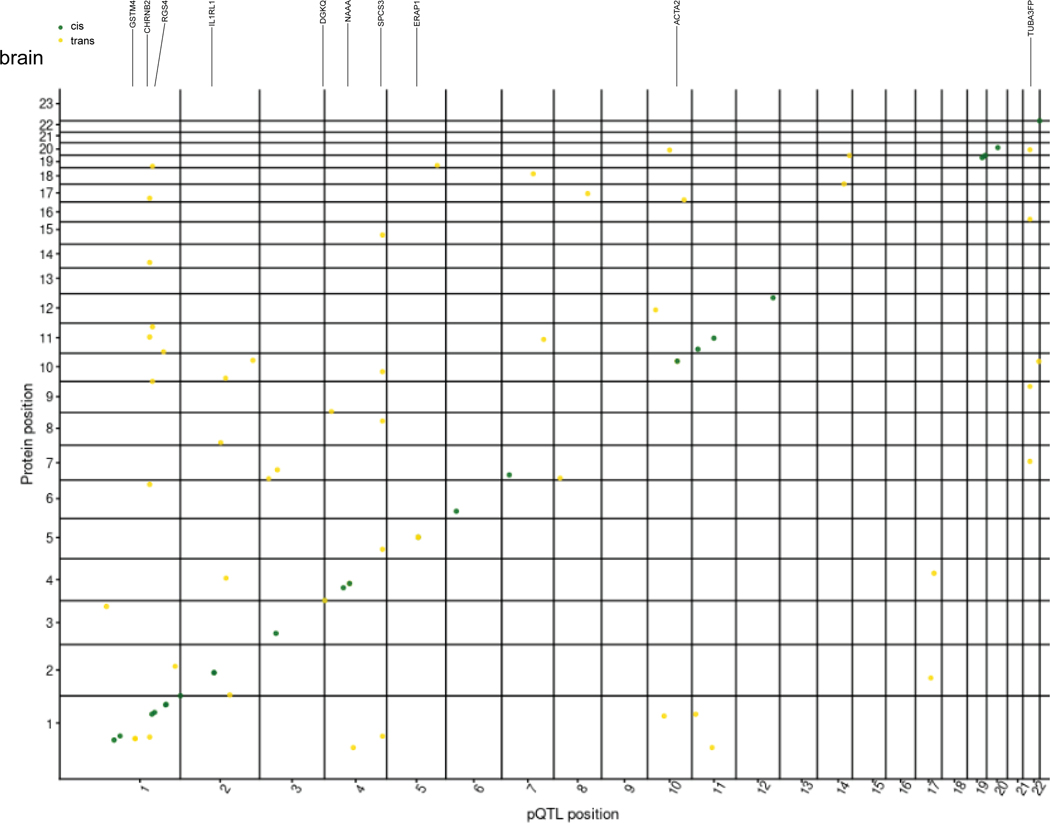
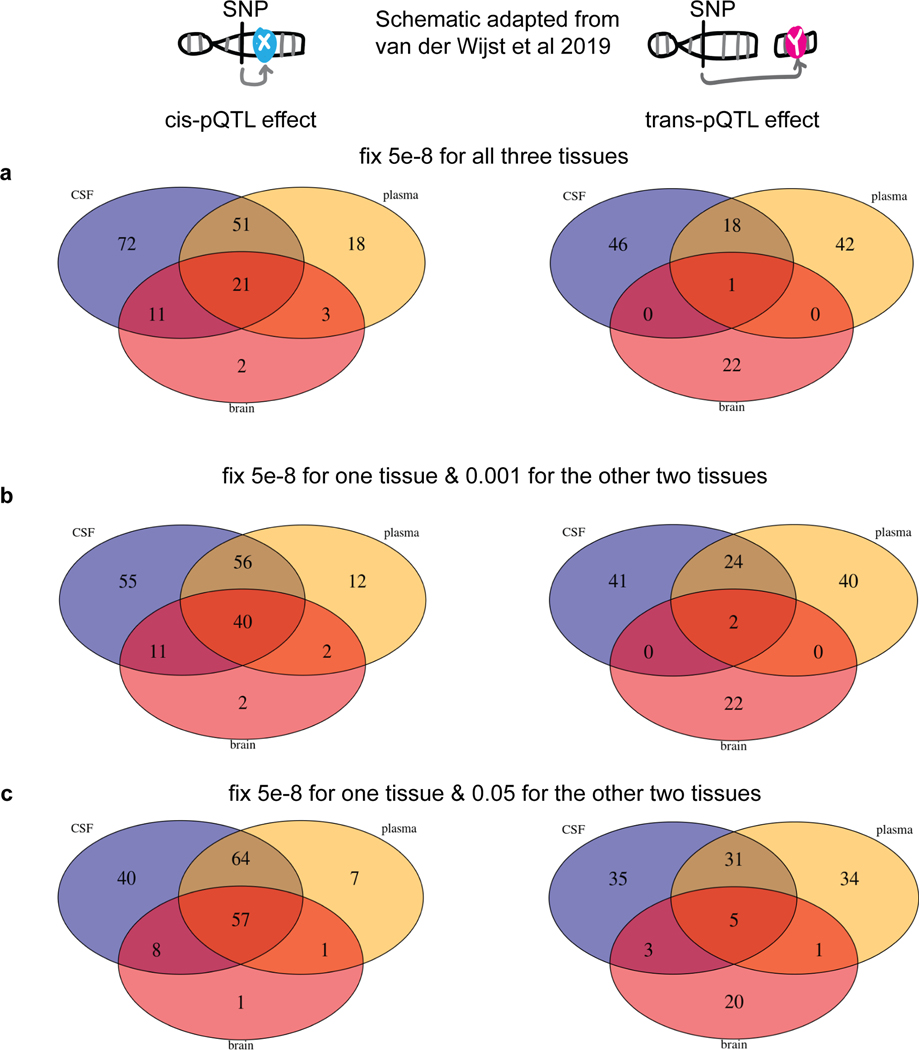
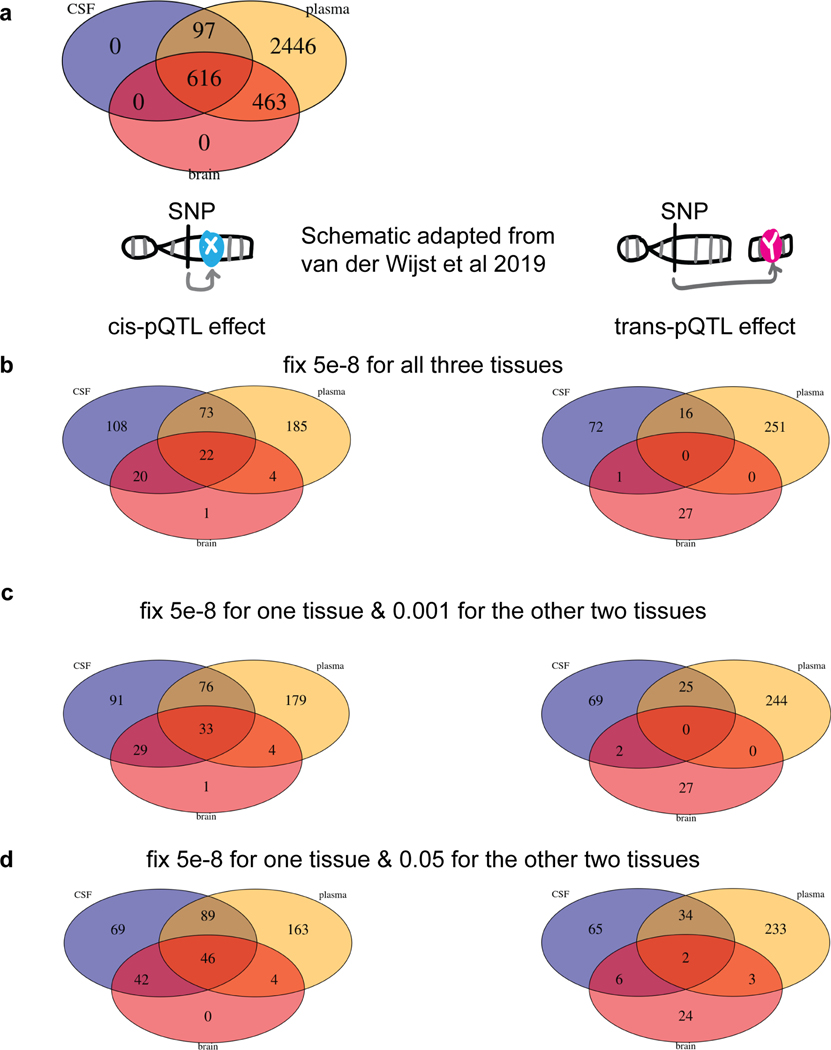
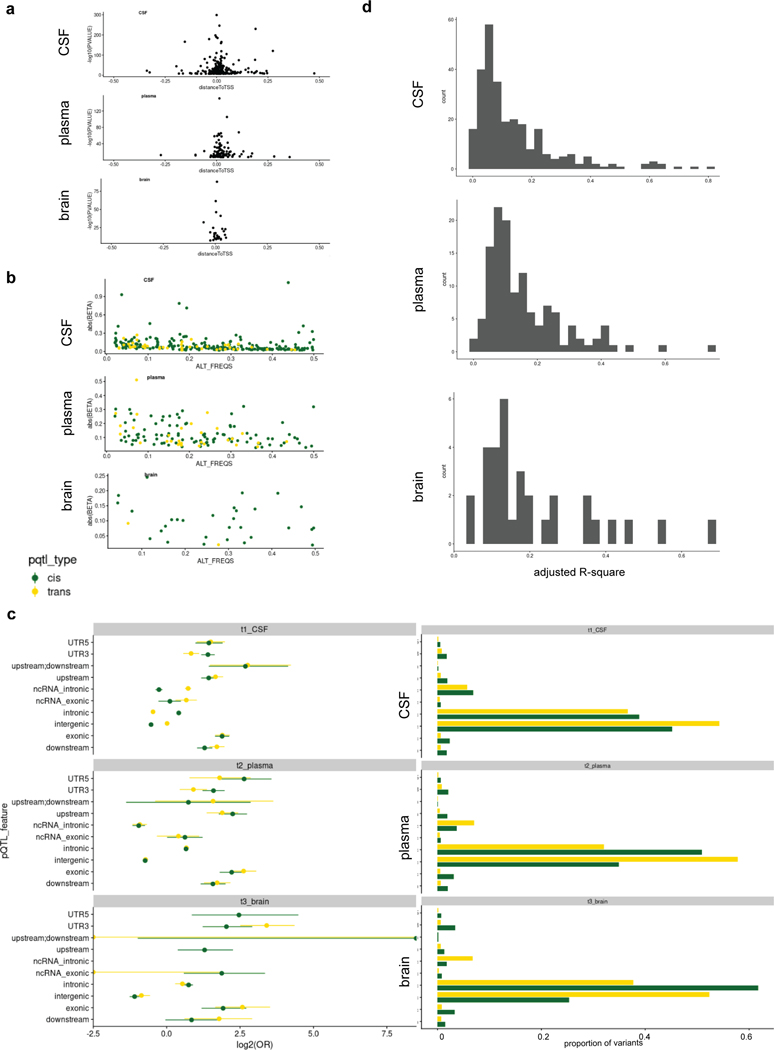
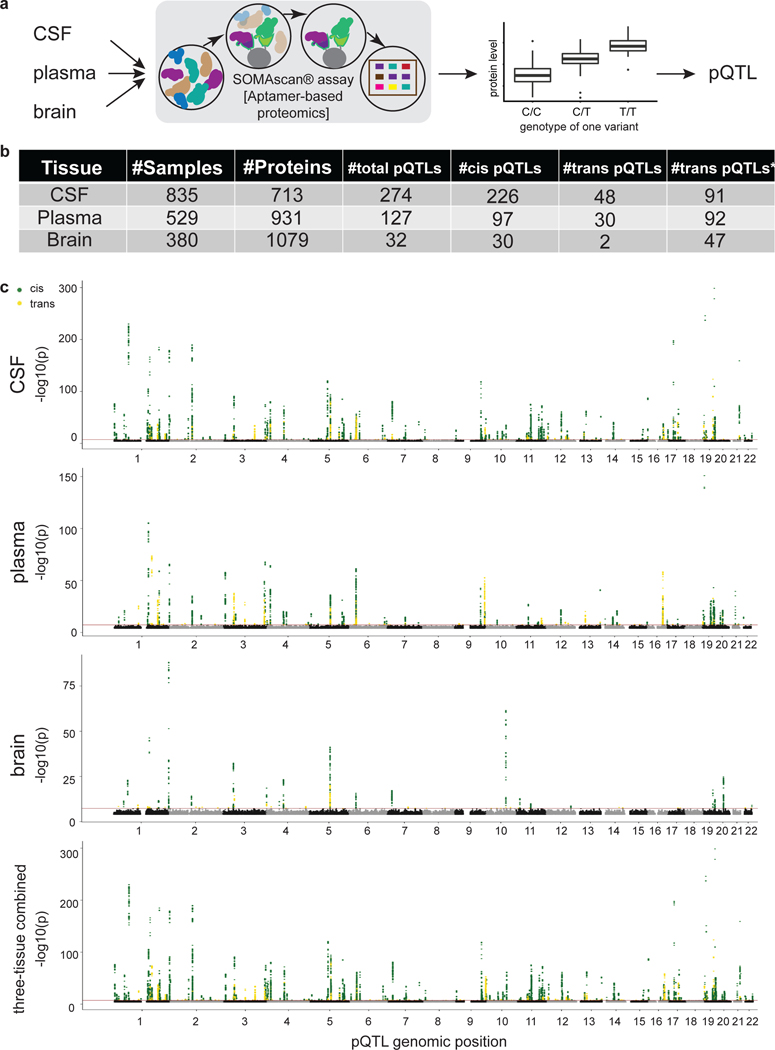
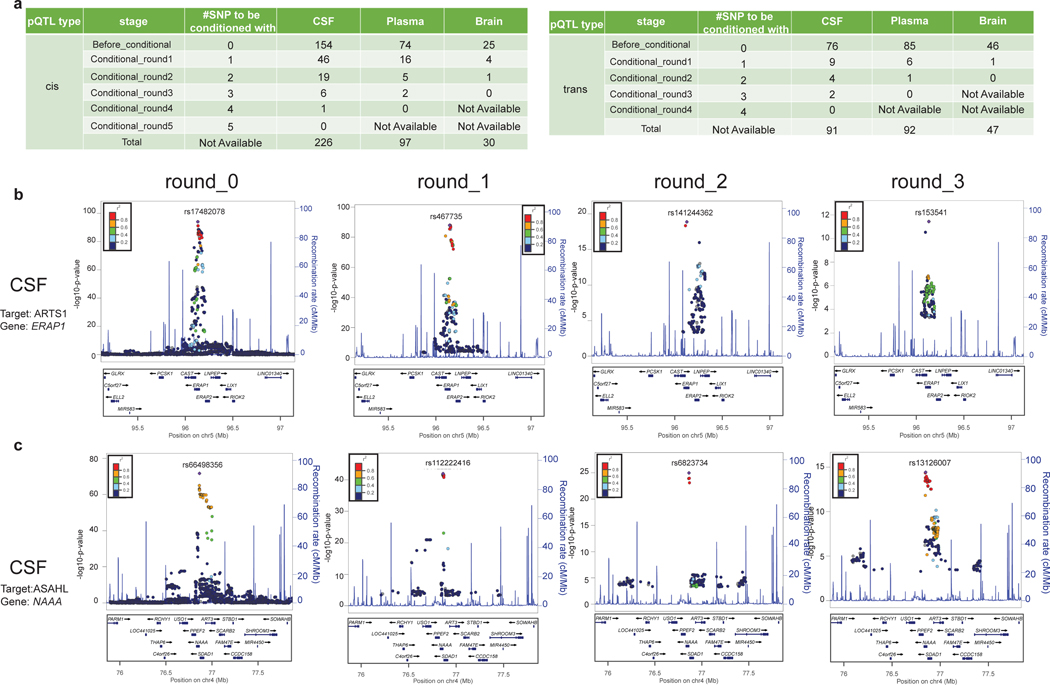
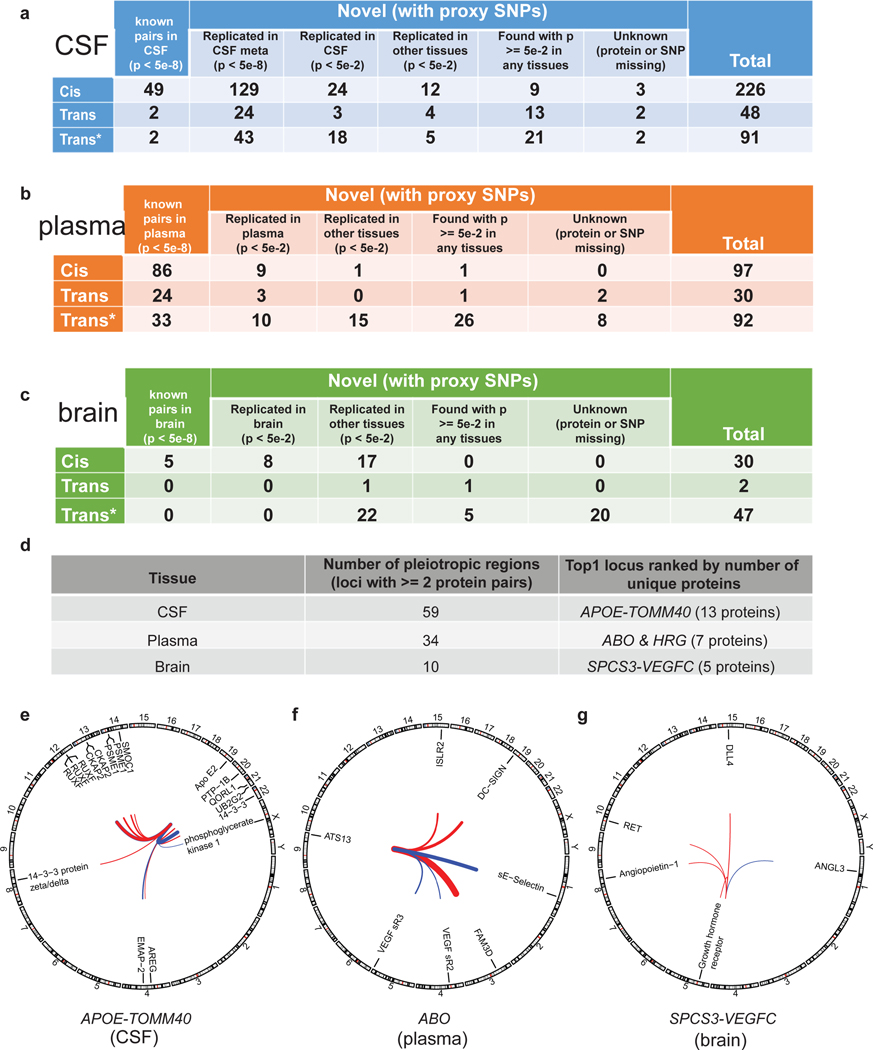
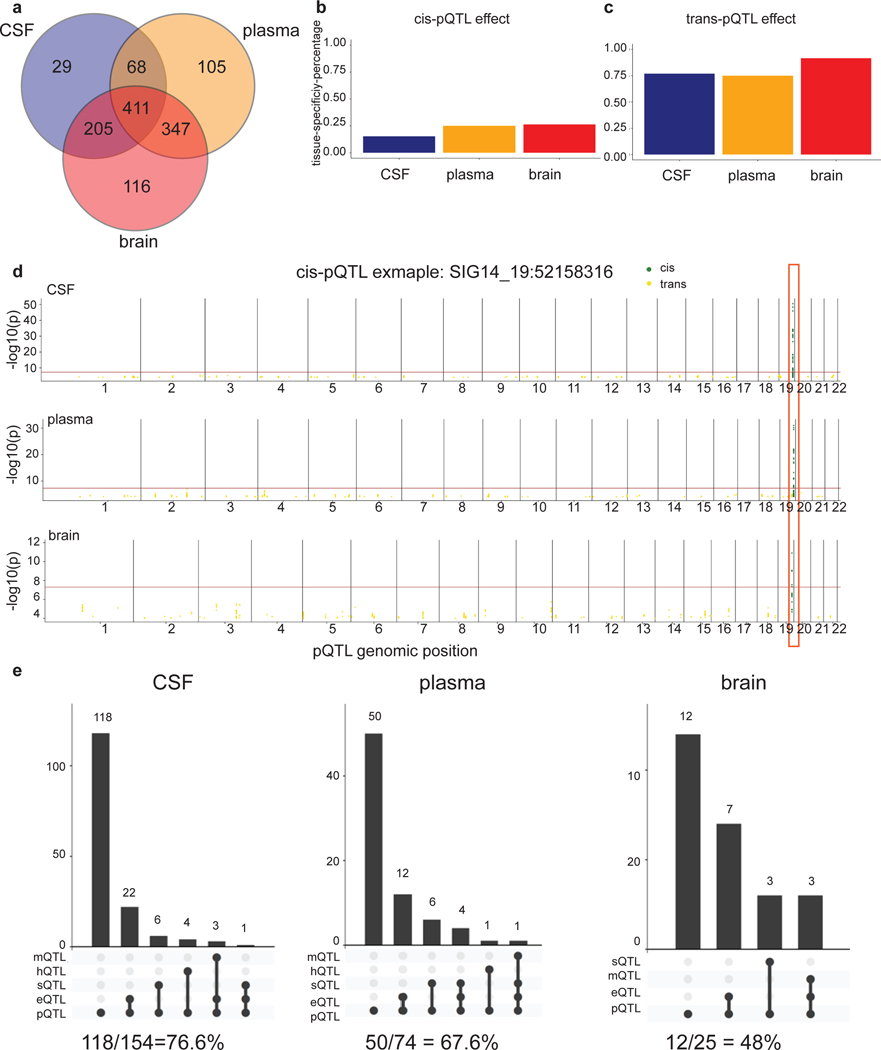
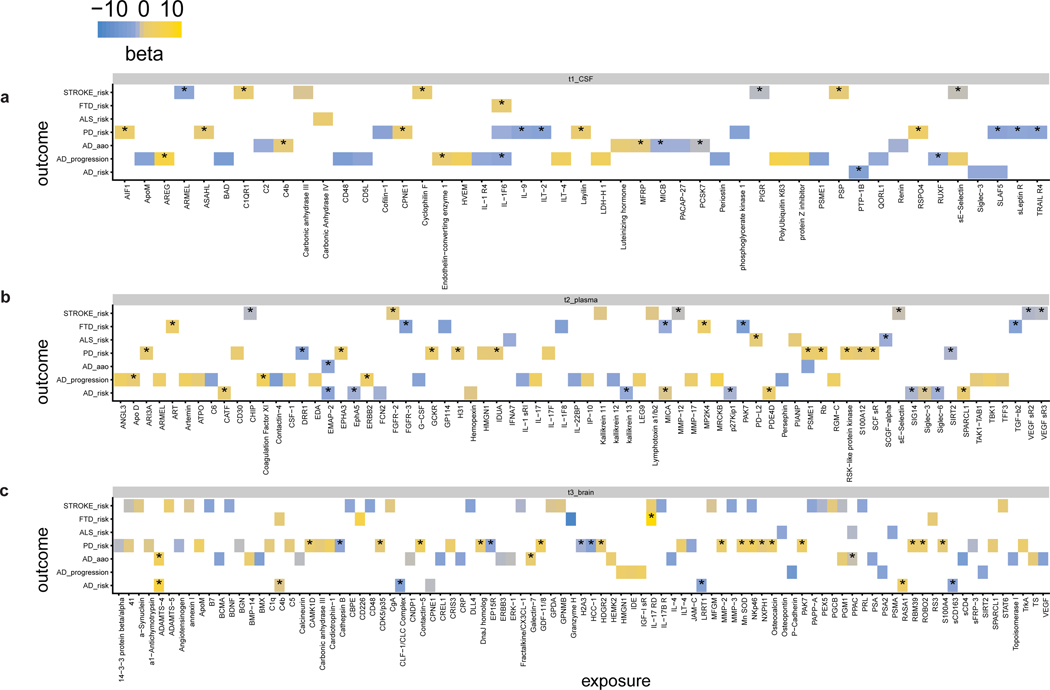
Understanding the tissue-specific genetic controls of protein levels is essential to uncover mechanisms of post-transcriptional gene regulation. In this study, we generated a genomic atlas of protein levels in three tissues relevant to neurological disorders (brain, cerebrospinal fluid and plasma) by profiling thousands of proteins from participants with and without Alzheimer's disease. We identified 274, 127 and 32 protein quantitative trait loci (pQTLs) for cerebrospinal fluid, plasma and brain, respectively. cis-pQTLs were more likely to be tissue shared, but trans-pQTLs tended to be tissue specific. Between 48.0% and 76.6% of pQTLs did not co-localize with expression, splicing, DNA methylation or histone acetylation QTLs. Using Mendelian randomization, we nominated proteins implicated in neurological diseases, including Alzheimer's disease, Parkinson's disease and stroke. This first multi-tissue study will be instrumental to map signals from genome-wide association studies onto functional genes, to discover pathways and to identify drug targets for neurological diseases.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
