Wilson's disease: Revisiting an old friend
- PMID: 34239699
- PMCID: PMC8239488
- DOI: 10.4254/wjh.v13.i6.634
Wilson's disease: Revisiting an old friend
Abstract
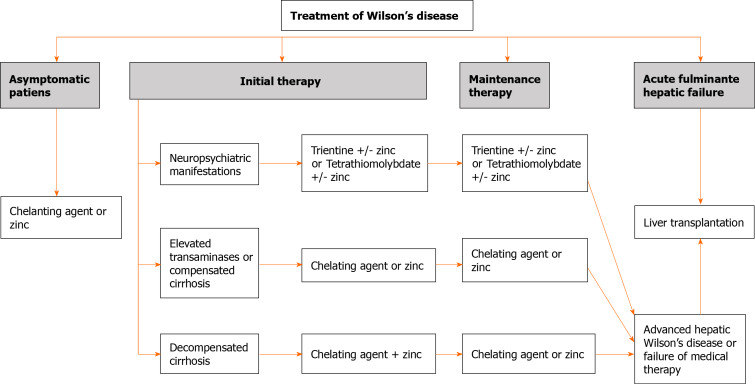
Wilson's disease (WD) is a rare condition caused by copper accumulation primarily in the liver and secondly in other organs, such as the central nervous system. It is a hereditary autosomal recessive disease caused by a deficiency in the ATP7B transporter. This protein facilitates the incorporation of copper into ceruloplasmin. More than 800 mutations associated with WD have been described. The onset of the disease frequently includes manifestations related to the liver (as chronic liver disease or acute liver failure) and neurological symptoms, although it can sometimes be asymptomatic. Despite it being more frequent in young people, WD has been described in all life stages. Due to its fatal prognosis, WD should be suspected in all patients with unexplained biochemical liver abnormalities or neurological or psychiatric symptoms. The diagnosis is established with a combination of clinical signs and tests, including the measurement of ceruloplasmin, urinary copper excretion, copper quantification in liver biopsy, or genetic assessment. The pharmacological therapies include chelating drugs, such as D-penicillamine or trientine, and zinc salts, which are able to change the natural history of the disease, increasing the survival of these patients. In some cases of end-stage liver disease or acute liver failure, liver transplantation must be an option to increase survival. In this narrative review, we offer an overview of WD, focusing on the importance of clinical suspicion, the correct diagnosis, and treatment.
Keywords: ATP7B; Ceruloplasmin; Chelator; Copper; Liver disease; Wilson´s disease.
©The Author(s) 2020. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Figures