

HIV-1 and SARS-CoV-2: Patterns in the evolution of two pandemic pathogens
- PMID: 34242582
- PMCID: PMC8173590
- DOI: 10.1016/j.chom.2021.05.012
HIV-1 and SARS-CoV-2: Patterns in the evolution of two pandemic pathogens
Abstract
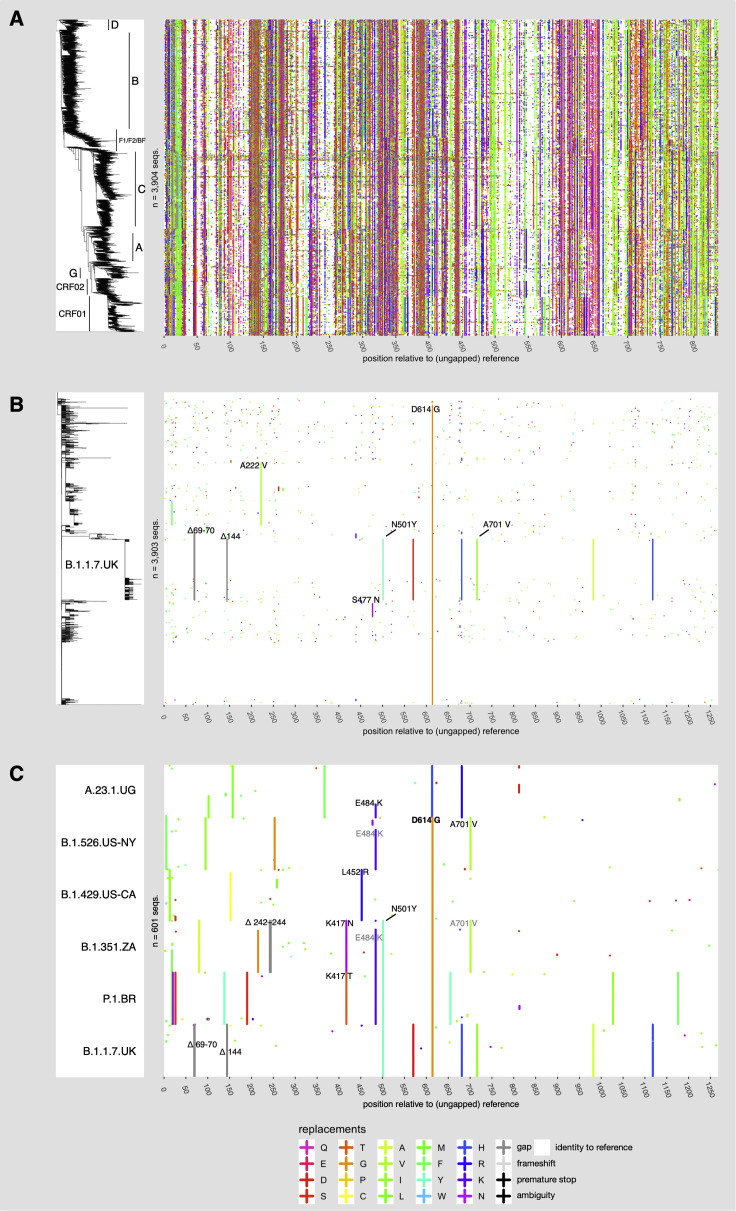
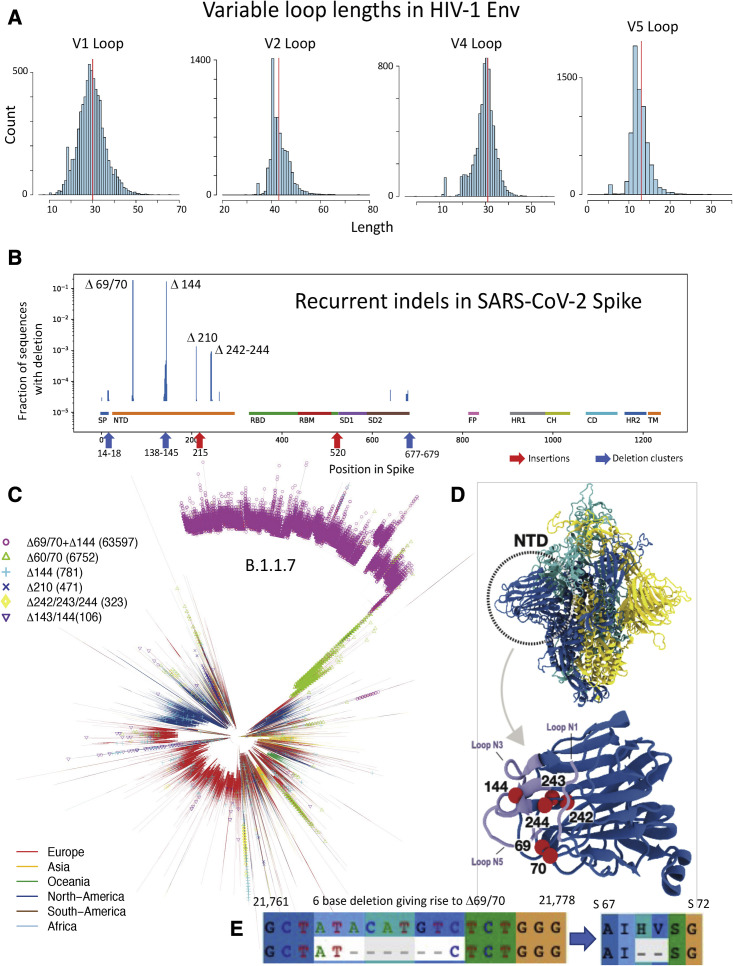
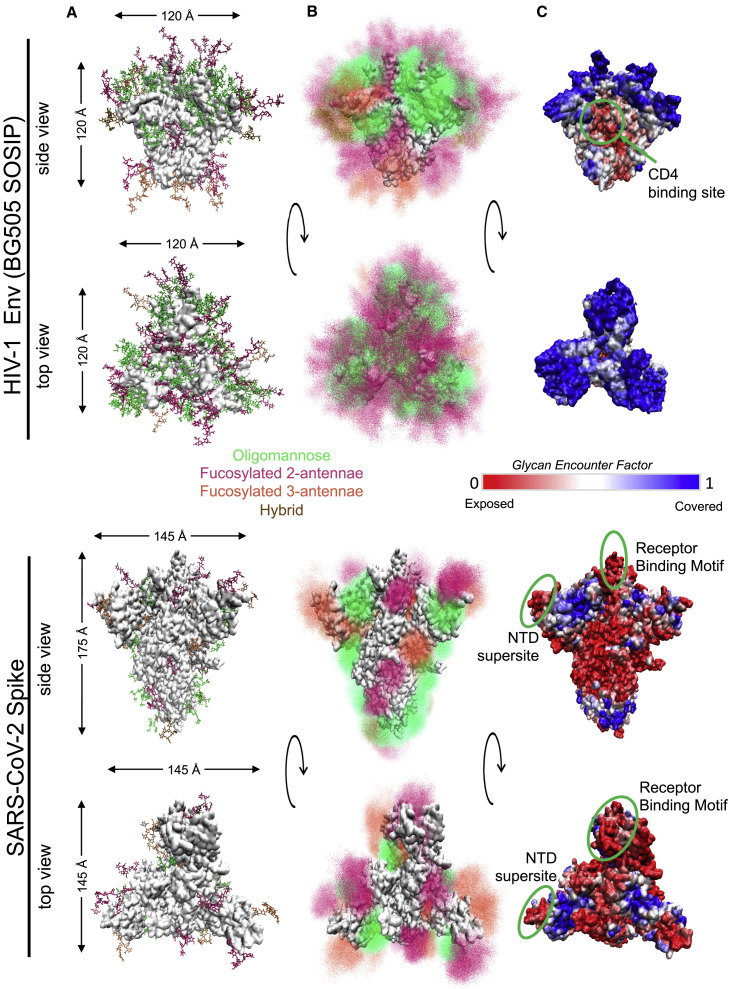
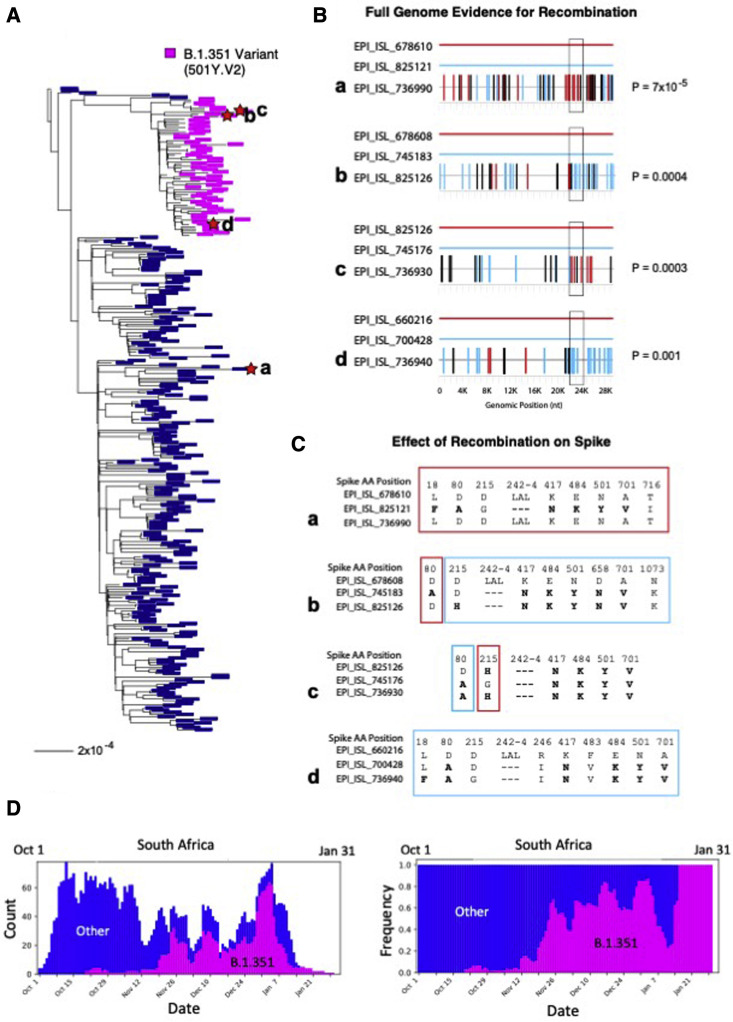
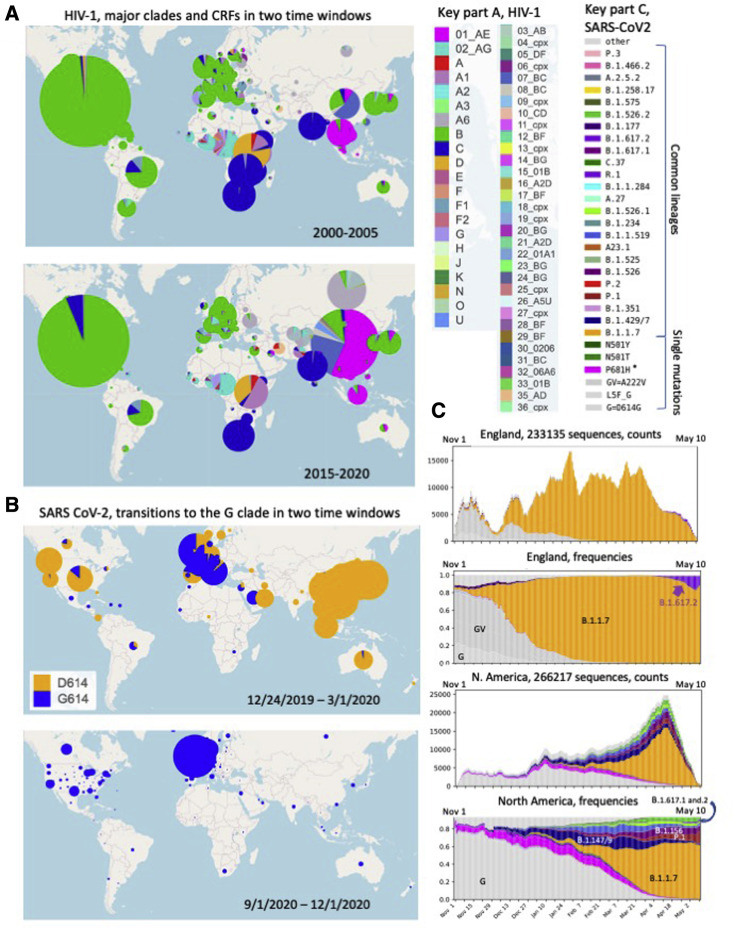
Humanity is currently facing the challenge of two devastating pandemics caused by two very different RNA viruses: HIV-1, which has been with us for decades, and SARS-CoV-2, which has swept the world in the course of a single year. The same evolutionary strategies that drive HIV-1 evolution are at play in SARS-CoV-2. Single nucleotide mutations, multi-base insertions and deletions, recombination, and variation in surface glycans all generate the variability that, guided by natural selection, enables both HIV-1's extraordinary diversity and SARS-CoV-2's slower pace of mutation accumulation. Even though SARS-CoV-2 diversity is more limited, recently emergent SARS-CoV-2 variants carry Spike mutations that have important phenotypic consequences in terms of both antibody resistance and enhanced infectivity. We review and compare how these mutational patterns manifest in these two distinct viruses to provide the variability that fuels their evolution by natural selection.
Keywords: HIV-1; SARS-CoV-2; evolution; glycosylation; immune escape; insertions and deletions; recombination.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests B.K., W.F., J.T., T.B., and S.G. have provisional patents and patents relating to vaccine design to address viral diversity as applied to HIV-1 and/or SARS-CoV-2.
Figures
References
-
- Arendrup M., Nielsen C., Hansen J.E., Pedersen C., Mathiesen L., Nielsen J.O. Autologous HIV-1 neutralizing antibodies: emergence of neutralization-resistant escape virus and subsequent development of escape virus neutralizing antibodies. J. Acquir. Immune Defic. Syndr. (1988) 1992;5:303–307. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
