

Characterizing nucleotide variation and expansion dynamics in human-specific variable number tandem repeats
- PMID: 34244228
- PMCID: PMC8327921
- DOI: 10.1101/gr.275560.121
Characterizing nucleotide variation and expansion dynamics in human-specific variable number tandem repeats
Abstract
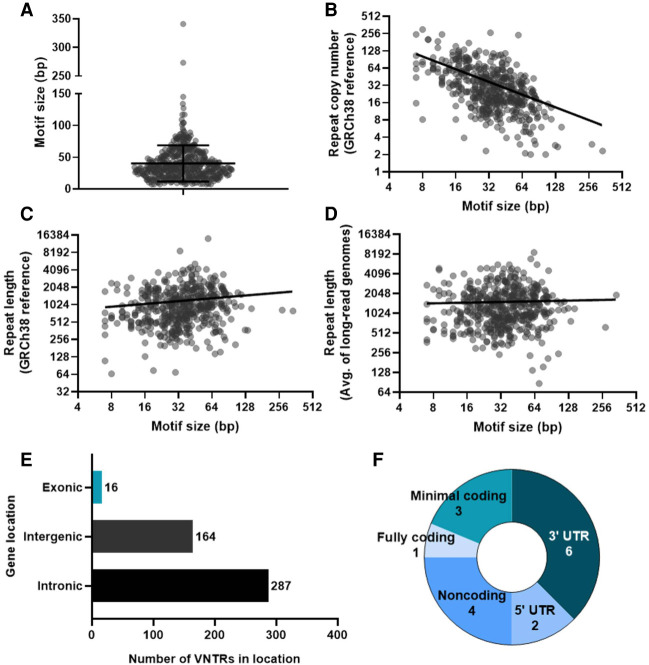
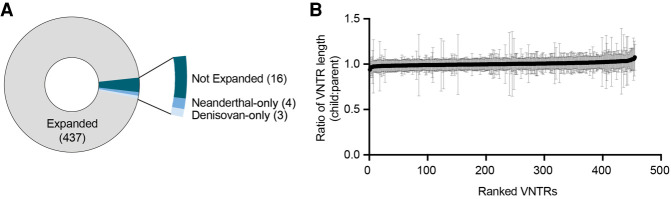
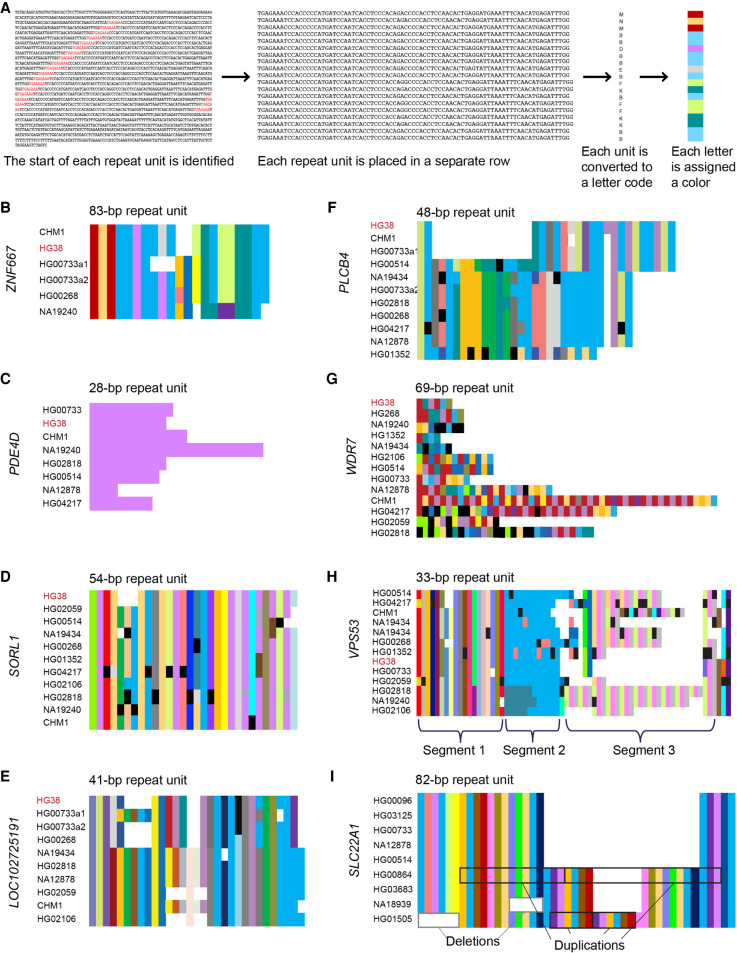
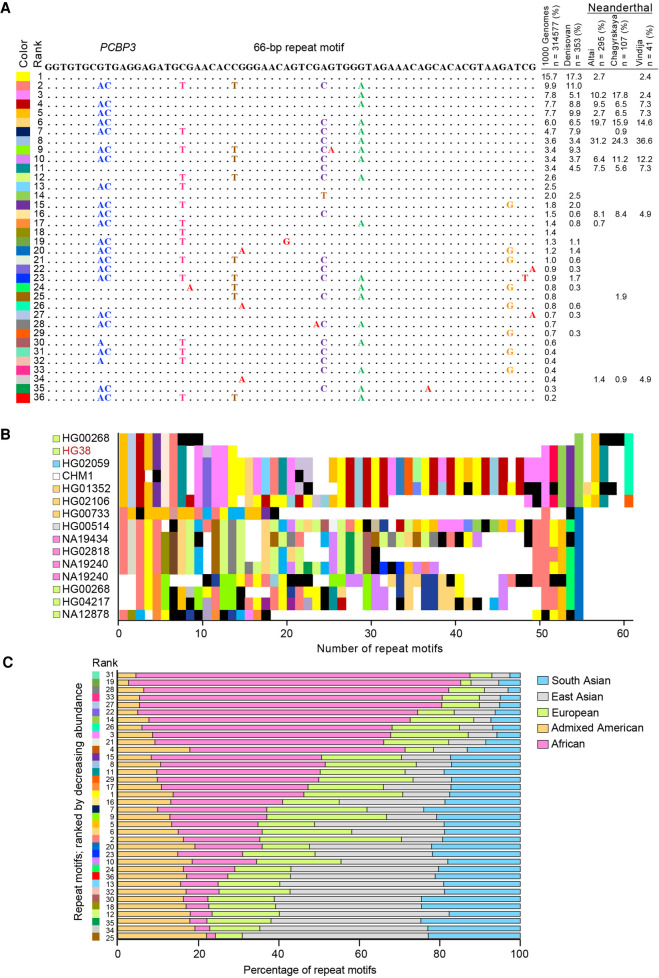
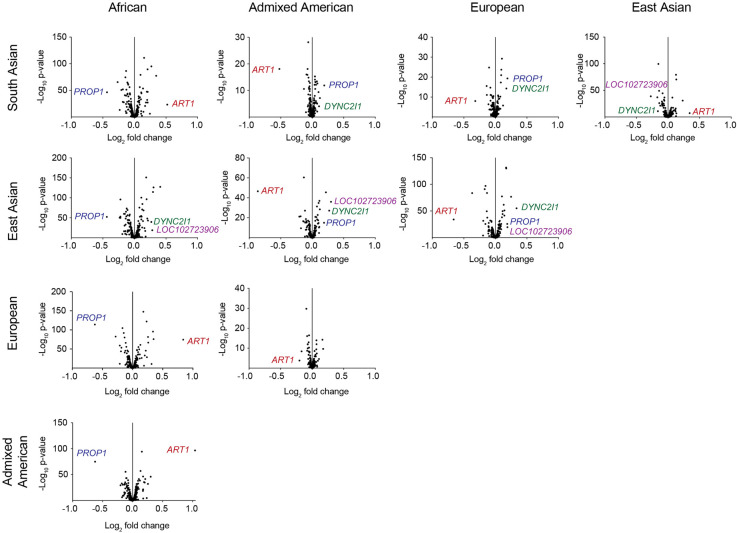
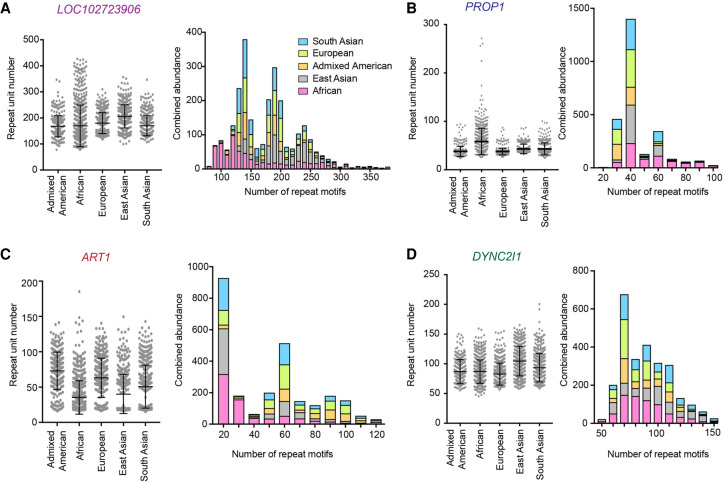
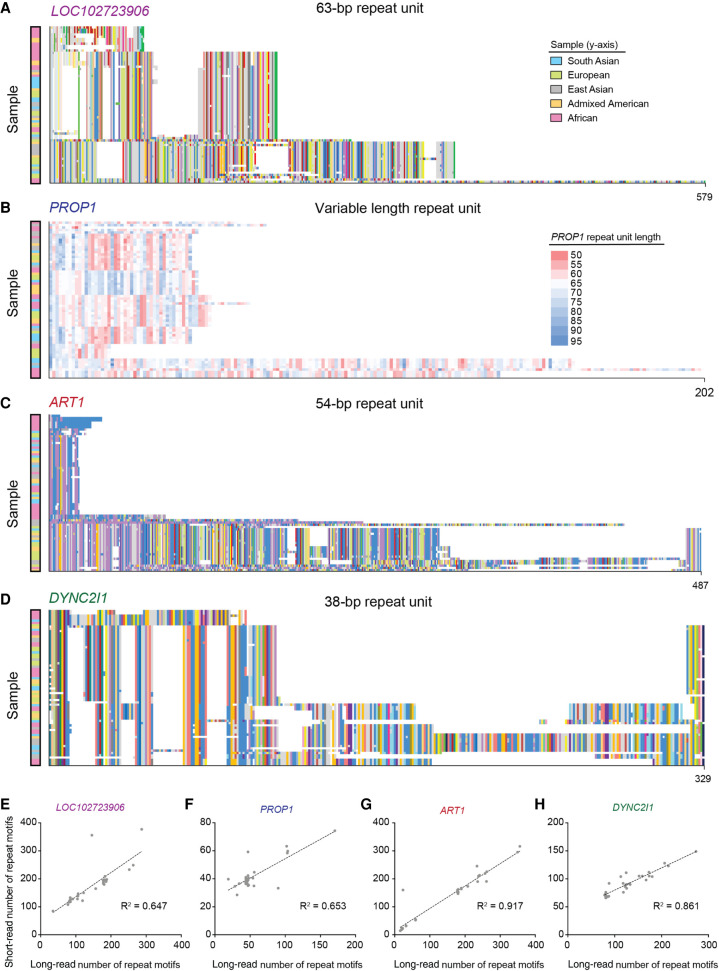
There are more than 55,000 variable number tandem repeats (VNTRs) in the human genome, notable for both their striking polymorphism and mutability. Despite their role in human evolution and genomic variation, they have yet to be studied collectively and in detail, partially owing to their large size, variability, and predominant location in noncoding regions. Here, we examine 467 VNTRs that are human-specific expansions, unique to one location in the genome, and not associated with retrotransposons. We leverage publicly available long-read genomes, including from the Human Genome Structural Variant Consortium, to ascertain the exact nucleotide composition of these VNTRs and compare their composition of alleles. We then confirm repeat unit composition in more than 3000 short-read samples from the 1000 Genomes Project. Our analysis reveals that these VNTRs contain highly structured repeat motif organization, modified by frequent deletion and duplication events. Although overall VNTR compositions tend to remain similar between 1000 Genomes Project superpopulations, we describe a notable exception with substantial differences in repeat composition (in PCBP3), as well as several VNTRs that are significantly different in length between superpopulations (in ART1, PROP1, DYNC2I1, and LOC102723906). We also observe that most of these VNTRs are expanded in archaic human genomes, yet remain stable in length between single generations. Collectively, our findings indicate that repeat motif variability, repeat composition, and repeat length are all informative modalities to consider when characterizing VNTRs and their contribution to genomic variation.
© 2021 Course et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources