

Genetic influences on hub connectivity of the human connectome
- PMID: 34244483
- PMCID: PMC8271018
- DOI: 10.1038/s41467-021-24306-2
Genetic influences on hub connectivity of the human connectome
Abstract
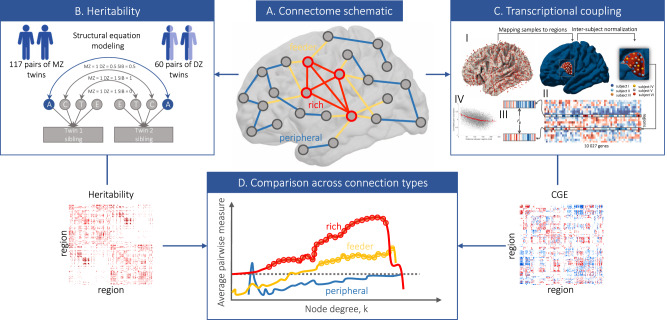
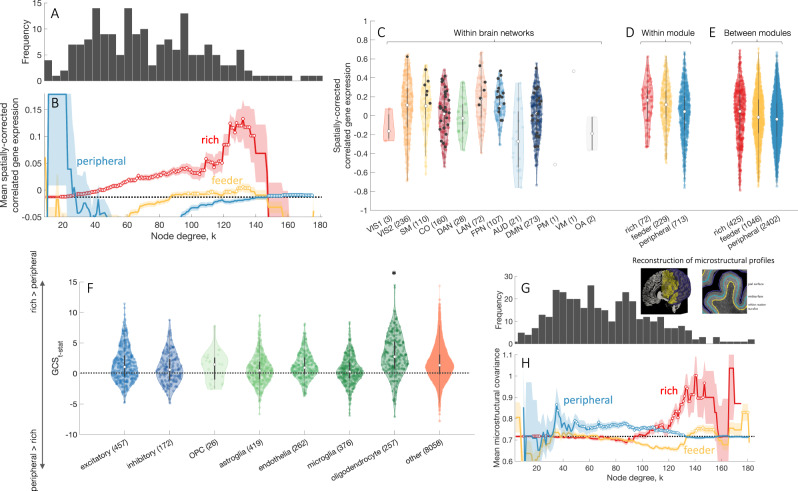
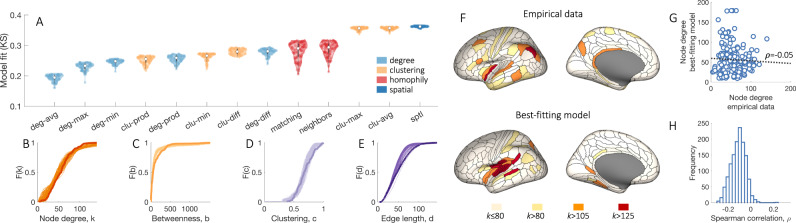
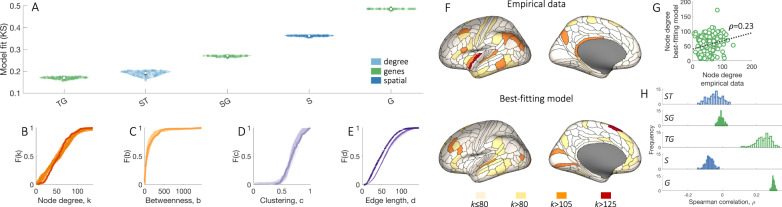
Brain network hubs are both highly connected and highly inter-connected, forming a critical communication backbone for coherent neural dynamics. The mechanisms driving this organization are poorly understood. Using diffusion-weighted magnetic resonance imaging in twins, we identify a major role for genes, showing that they preferentially influence connectivity strength between network hubs of the human connectome. Using transcriptomic atlas data, we show that connected hubs demonstrate tight coupling of transcriptional activity related to metabolic and cytoarchitectonic similarity. Finally, comparing over thirteen generative models of network growth, we show that purely stochastic processes cannot explain the precise wiring patterns of hubs, and that model performance can be improved by incorporating genetic constraints. Our findings indicate that genes play a strong and preferential role in shaping the functionally valuable, metabolically costly connections between connectome hubs.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Fornito, A., Zalesky, A. & Bullmore, E. Fundamentals of Brain Network Analysis (Academic Press, 2016).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
