

Biallelic variants in PCDHGC4 cause a novel neurodevelopmental syndrome with progressive microcephaly, seizures, and joint anomalies
- PMID: 34244665
- PMCID: PMC8553613
- DOI: 10.1038/s41436-021-01260-4
Biallelic variants in PCDHGC4 cause a novel neurodevelopmental syndrome with progressive microcephaly, seizures, and joint anomalies
Abstract
Purpose: We aimed to define a novel autosomal recessive neurodevelopmental disorder, characterize its clinical features, and identify the underlying genetic cause for this condition.
Methods: We performed a detailed clinical characterization of 19 individuals from nine unrelated, consanguineous families with a neurodevelopmental disorder. We used genome/exome sequencing approaches, linkage and cosegregation analyses to identify disease-causing variants, and we performed three-dimensional molecular in silico analysis to predict causality of variants where applicable.
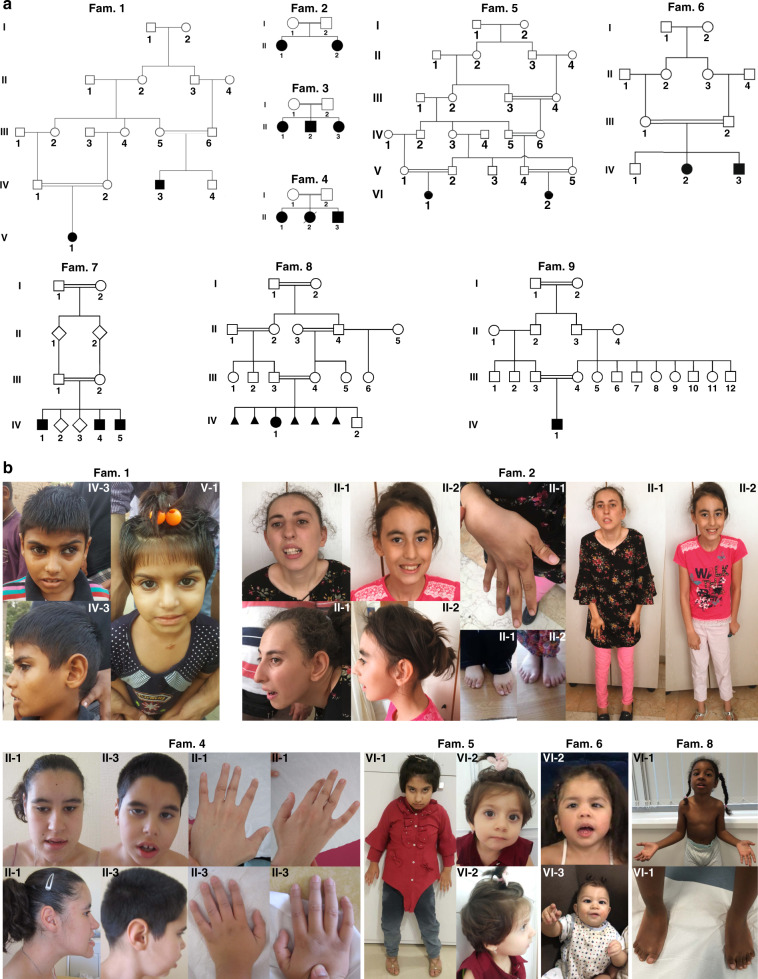
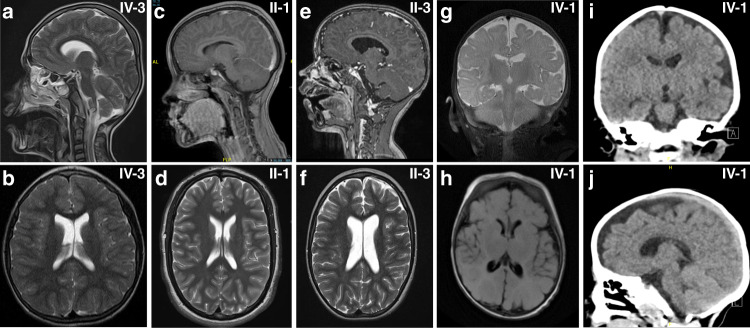
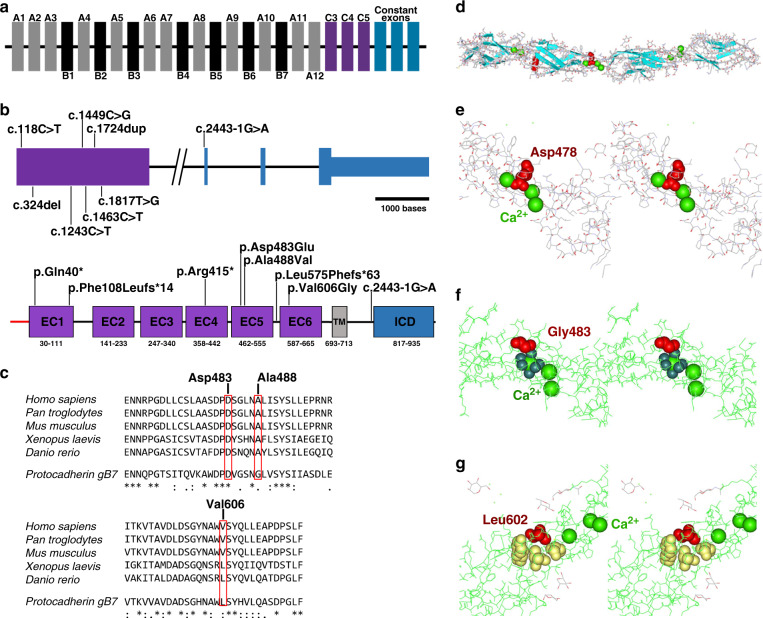
Results: In all affected individuals who presented with a neurodevelopmental syndrome with progressive microcephaly, seizures, and intellectual disability we identified biallelic disease-causing variants in Protocadherin-gamma-C4 (PCDHGC4). Five variants were predicted to induce premature protein truncation leading to a loss of PCDHGC4 function. The three detected missense variants were located in extracellular cadherin (EC) domains EC5 and EC6 of PCDHGC4, and in silico analysis of the affected residues showed that two of these substitutions were predicted to influence the Ca2+-binding affinity, which is essential for multimerization of the protein, whereas the third missense variant directly influenced the cis-dimerization interface of PCDHGC4.
Conclusion: We show that biallelic variants in PCDHGC4 are causing a novel autosomal recessive neurodevelopmental disorder and link PCDHGC4 as a member of the clustered PCDH family to a Mendelian disorder in humans.
© 2021. The Author(s).
Conflict of interest statement
V.K. and C.B. are employees of Centogene GmbH (Rostock, Germany). The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
