

Smart-RRBS for single-cell methylome and transcriptome analysis
- PMID: 34244697
- PMCID: PMC8672372
- DOI: 10.1038/s41596-021-00571-9
Smart-RRBS for single-cell methylome and transcriptome analysis
Abstract
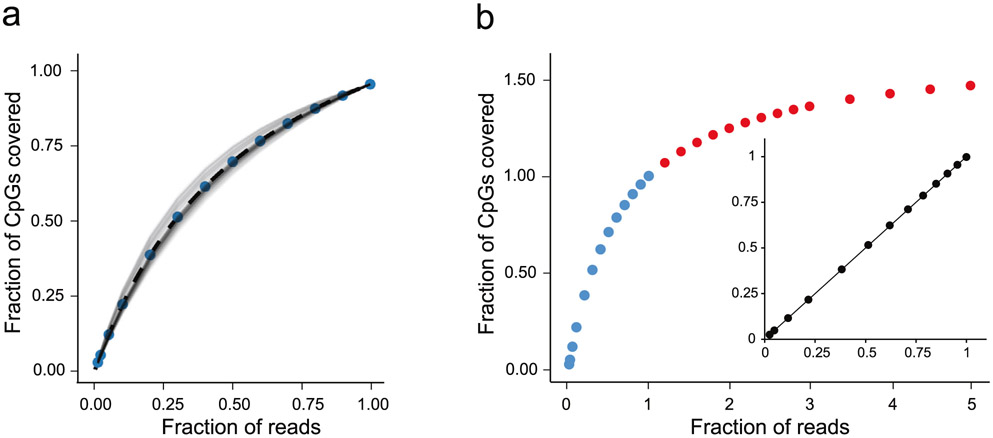
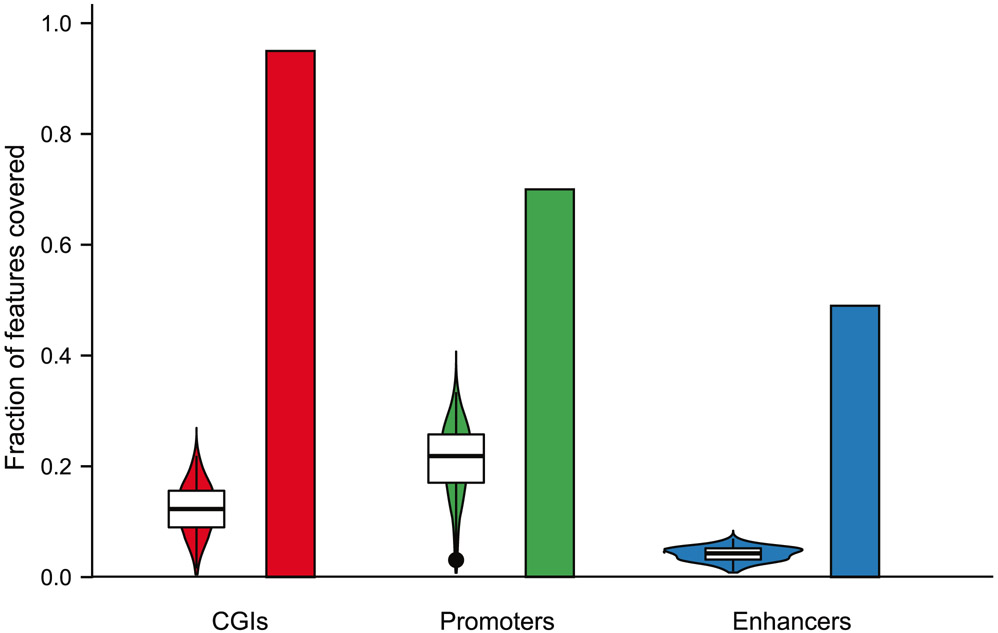
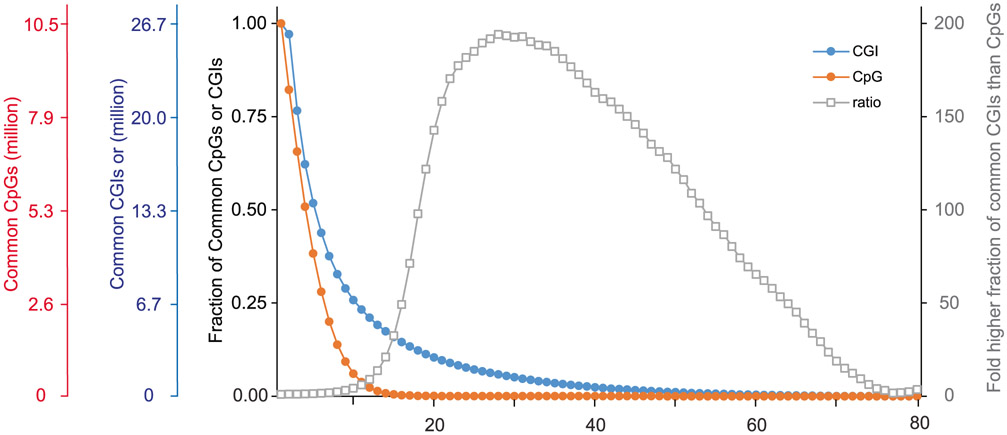
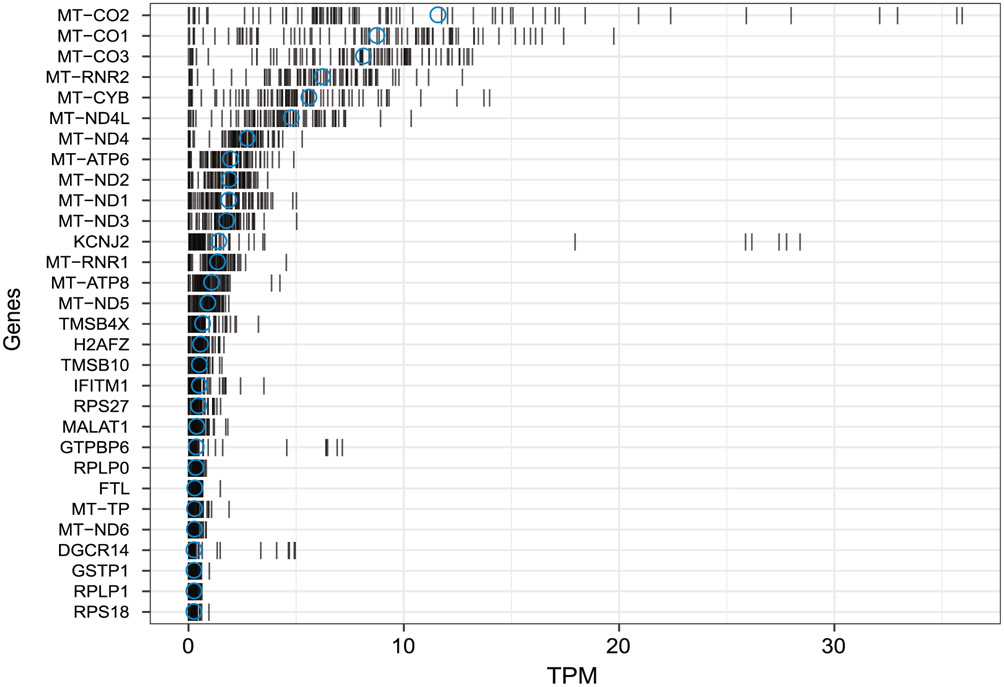
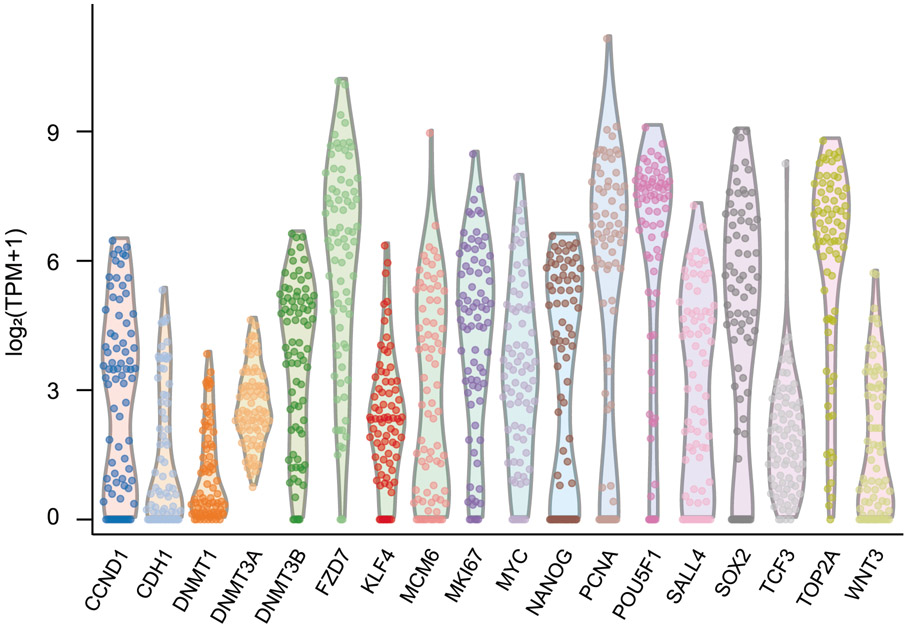
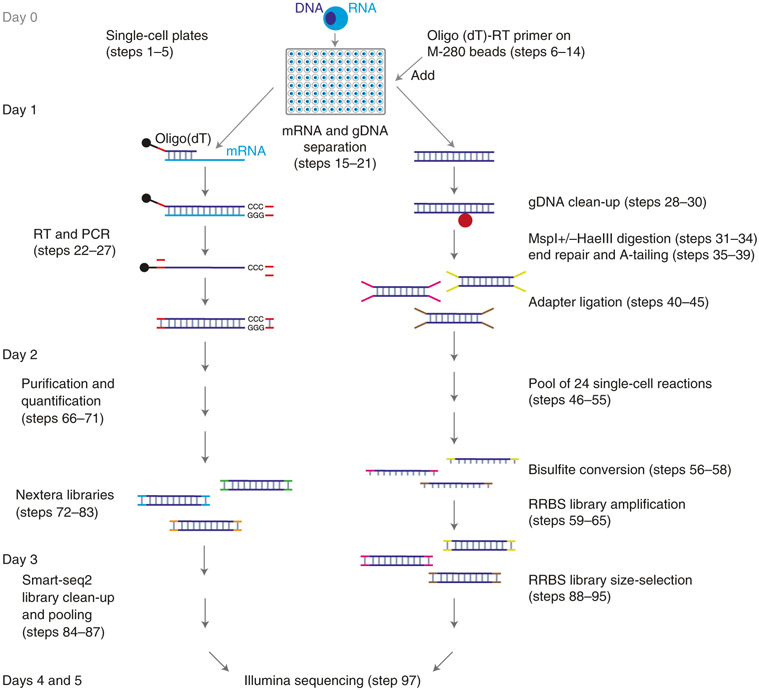
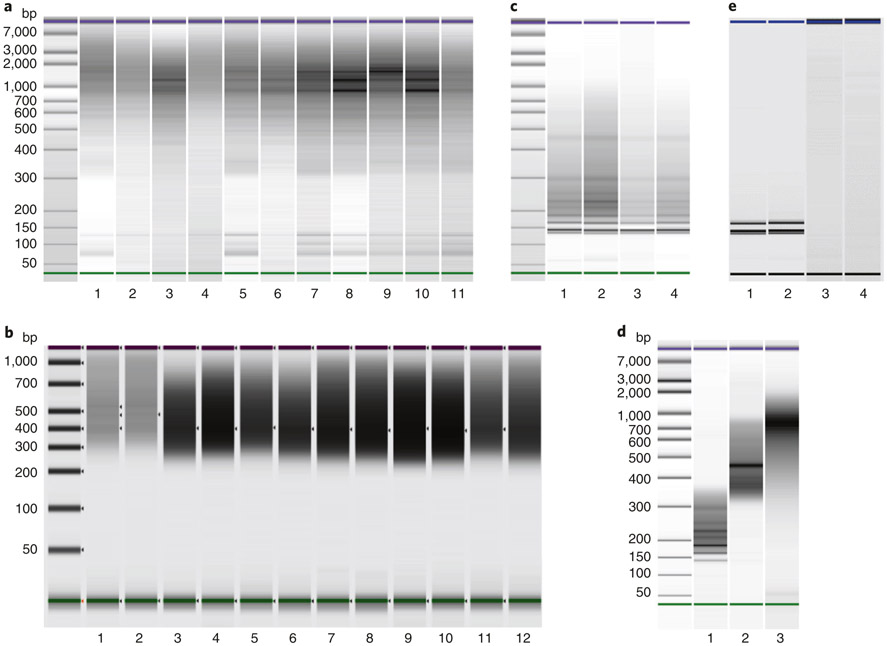
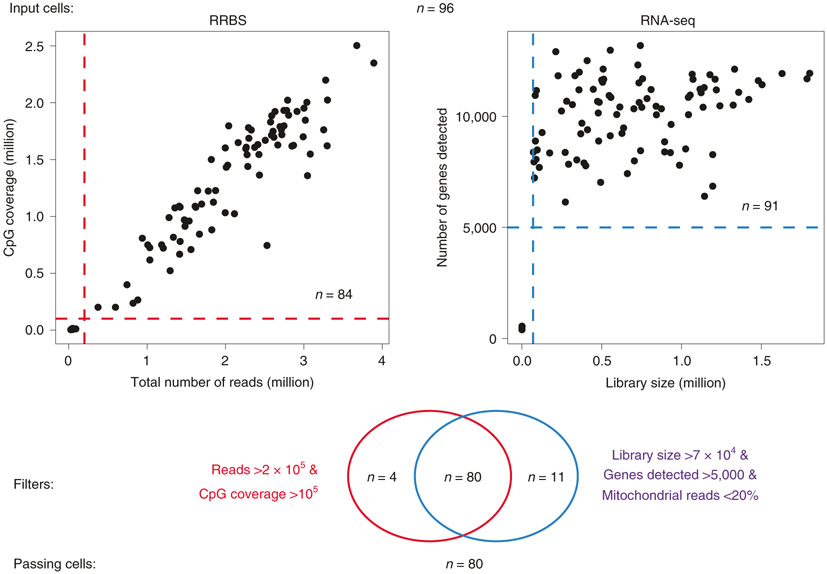
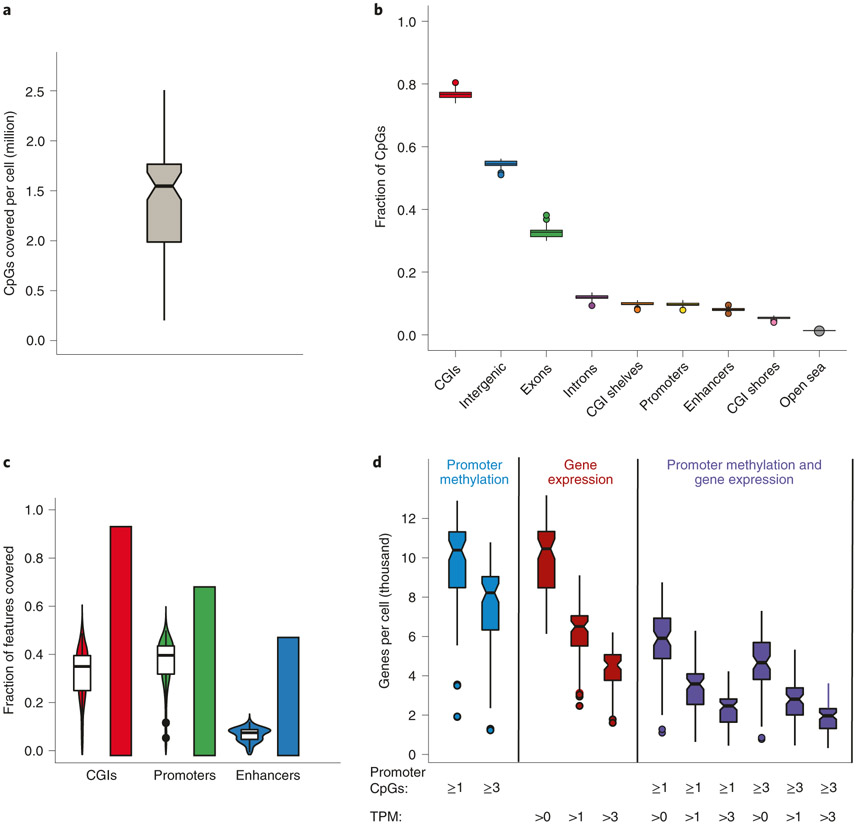
The integration of DNA methylation and transcriptional state within single cells is of broad interest. Several single-cell dual- and multi-omics approaches have been reported that enable further investigation into cellular heterogeneity, including the discovery and in-depth study of rare cell populations. Such analyses will continue to provide important mechanistic insights into the regulatory consequences of epigenetic modifications. We recently reported a new method for profiling the DNA methylome and transcriptome from the same single cells in a cancer research study. Here, we present details of the protocol and provide guidance on its utility. Our Smart-RRBS (reduced representation bisulfite sequencing) protocol combines Smart-seq2 and RRBS and entails physically separating mRNA from the genomic DNA. It generates paired epigenetic promoter and RNA-expression measurements for ~24% of protein-coding genes in a typical single cell. It also works for micro-dissected tissue samples comprising hundreds of cells. The protocol, excluding flow sorting of cells and sequencing, takes ~3 d to process up to 192 samples manually. It requires basic molecular biology expertise and laboratory equipment, including a PCR workstation with UV sterilization, a DNA fluorometer and a microfluidic electrophoresis system.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
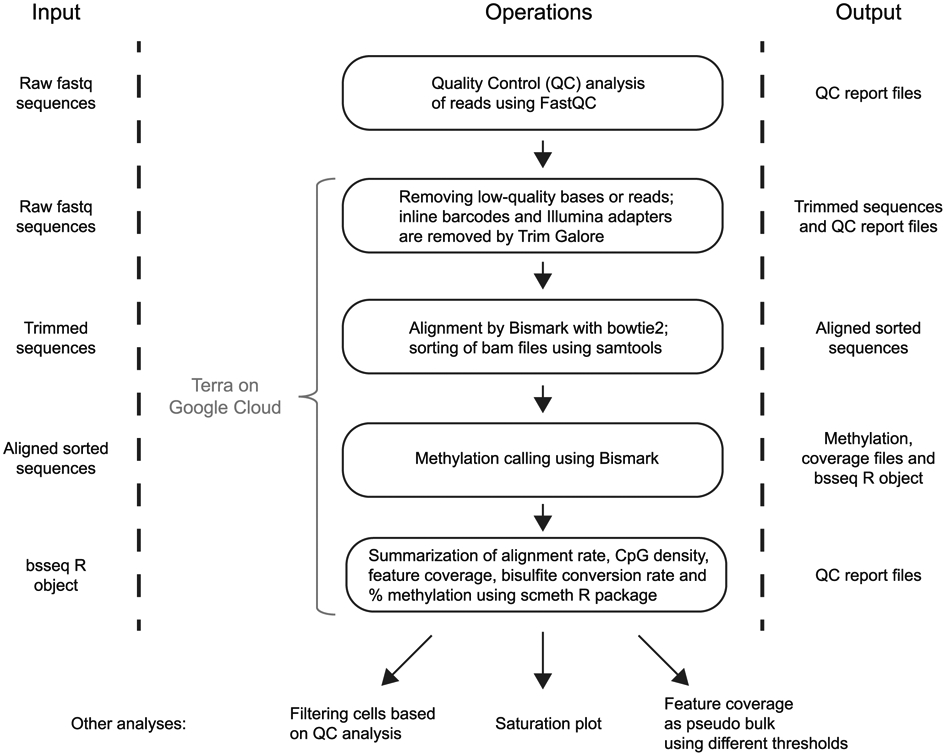
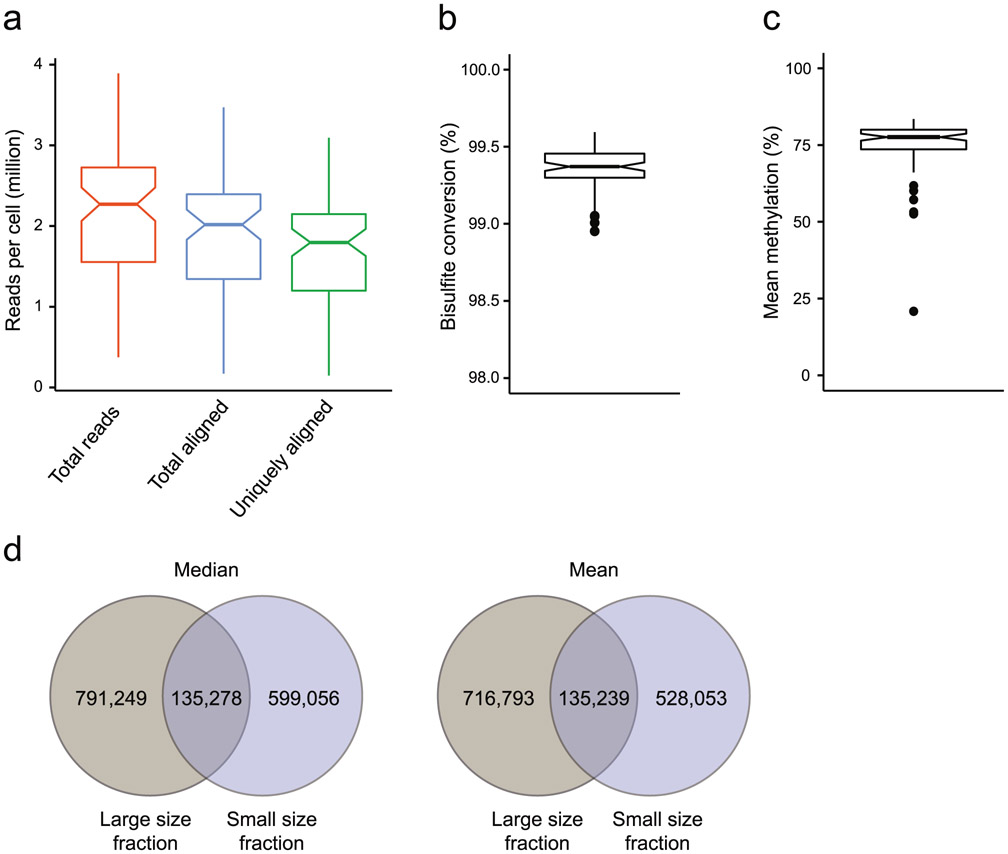
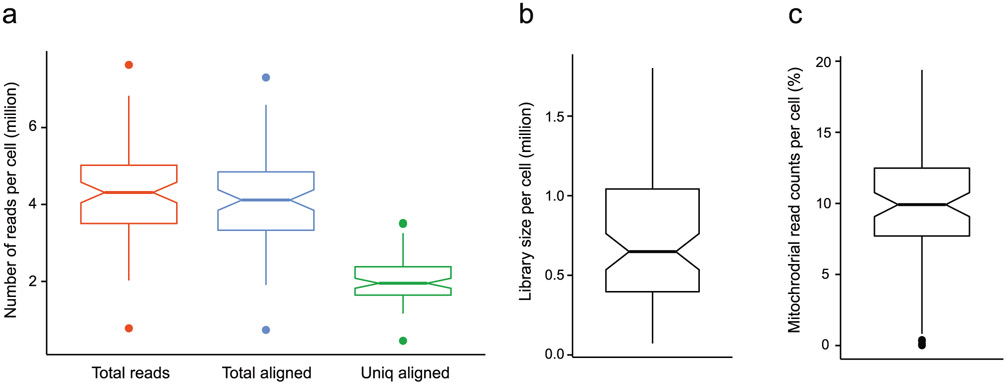
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
