

Reference Transcriptomes of Porcine Peripheral Immune Cells Created Through Bulk and Single-Cell RNA Sequencing
- PMID: 34249103
- PMCID: PMC8261551
- DOI: 10.3389/fgene.2021.689406
Reference Transcriptomes of Porcine Peripheral Immune Cells Created Through Bulk and Single-Cell RNA Sequencing
Abstract
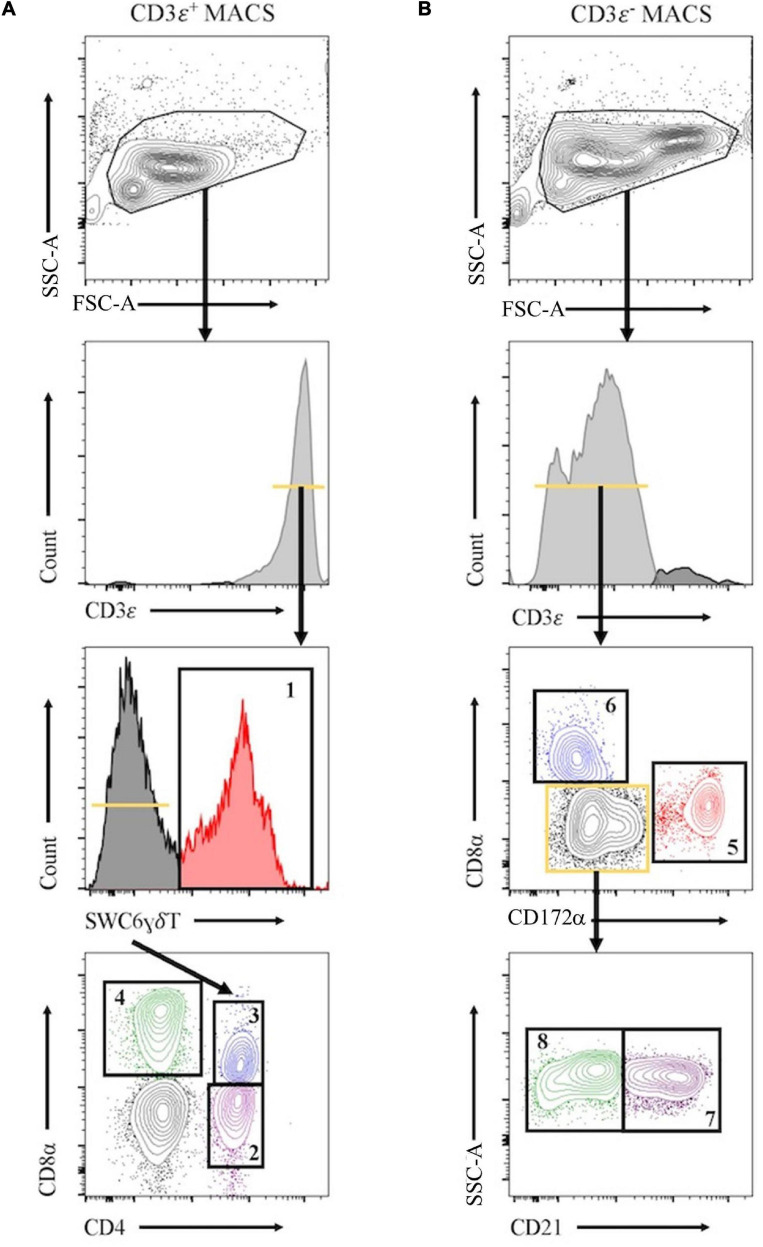
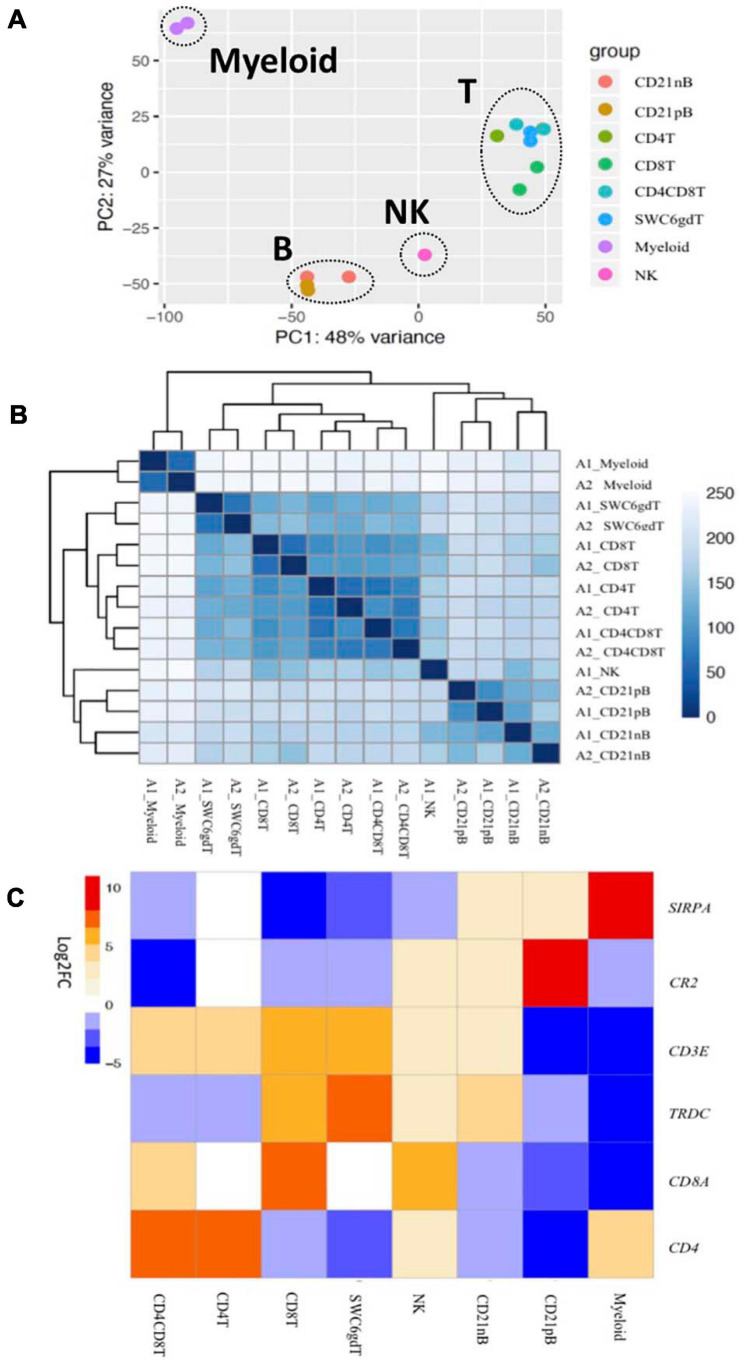
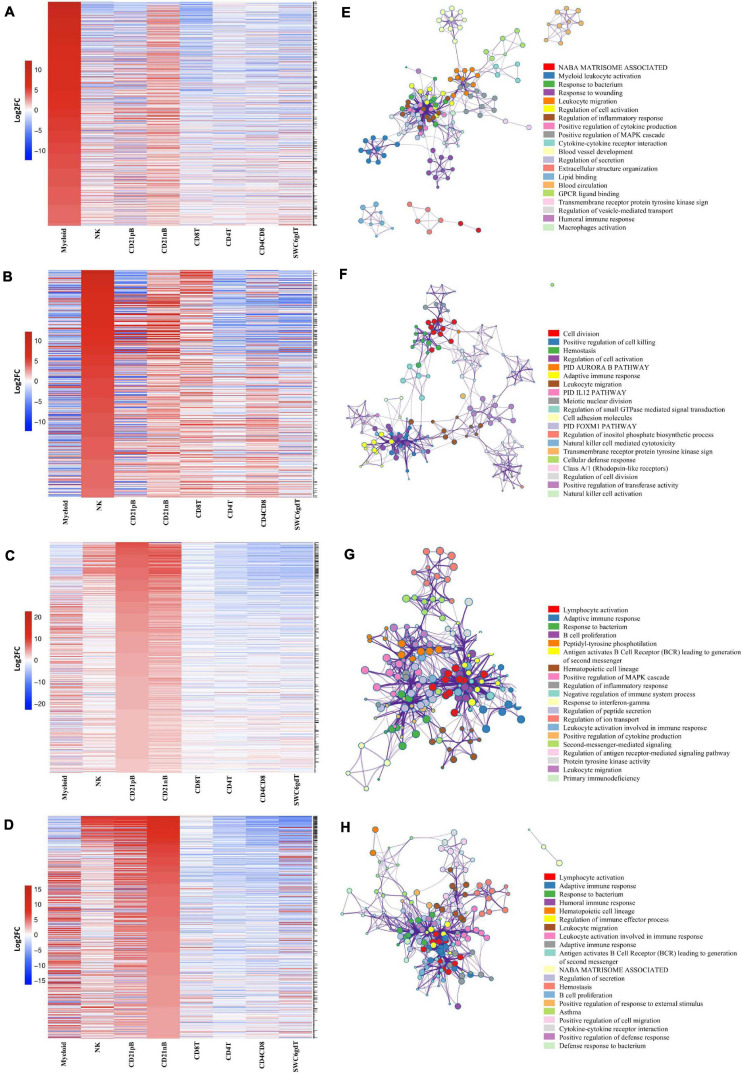
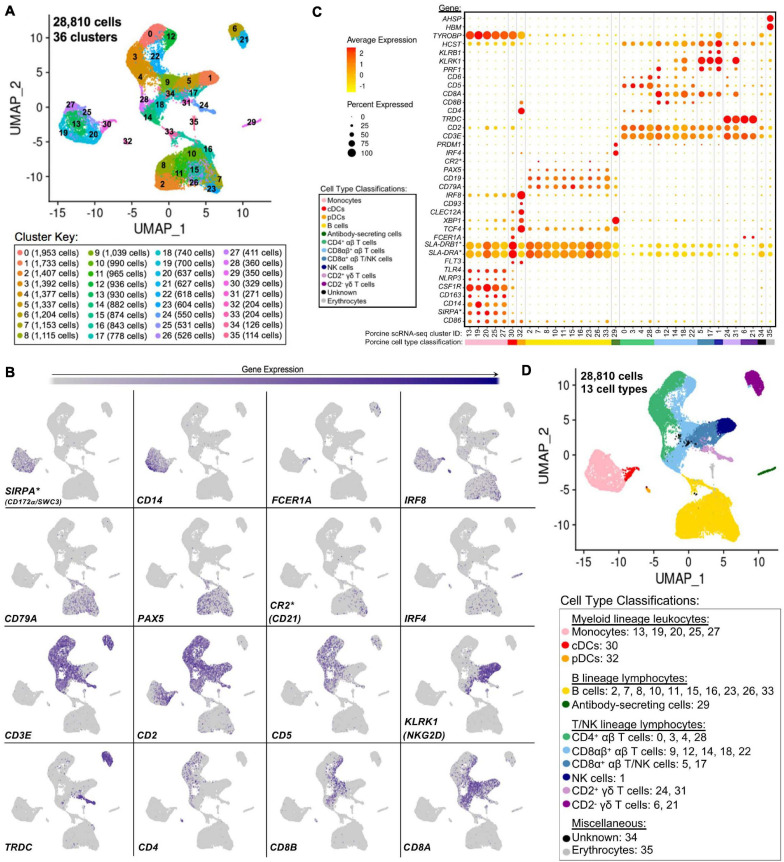
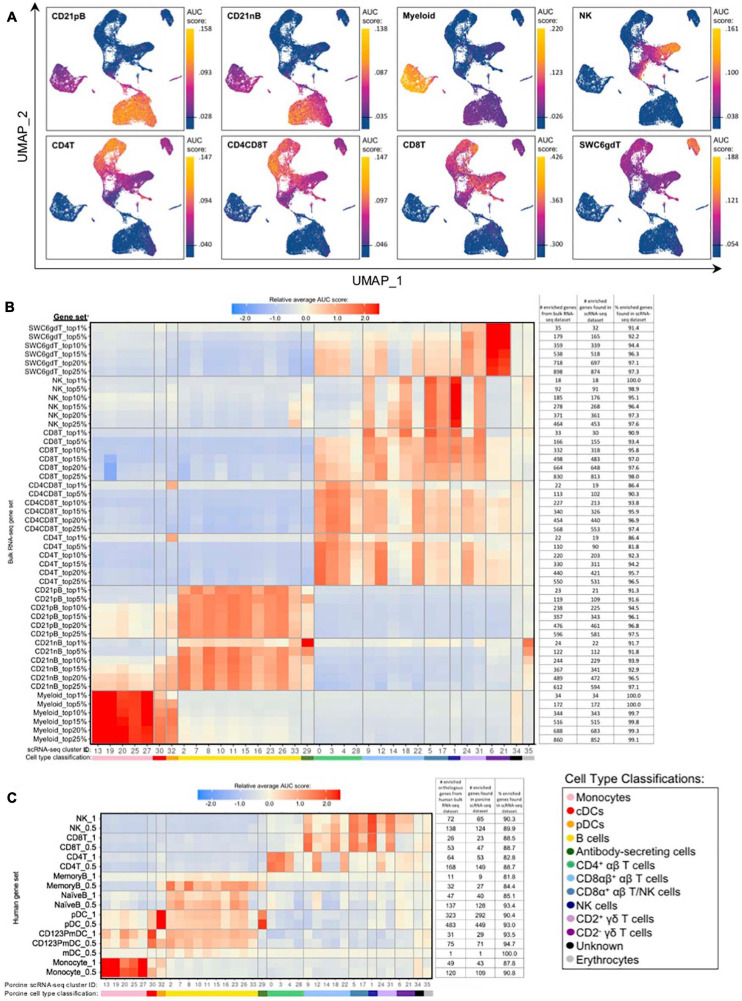
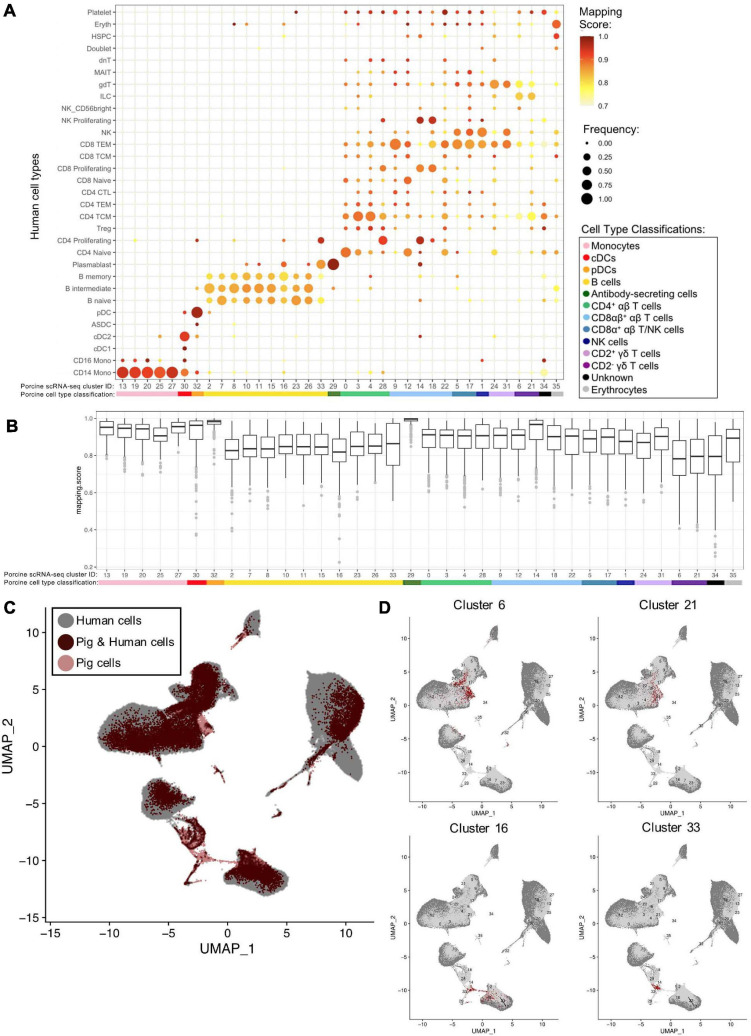
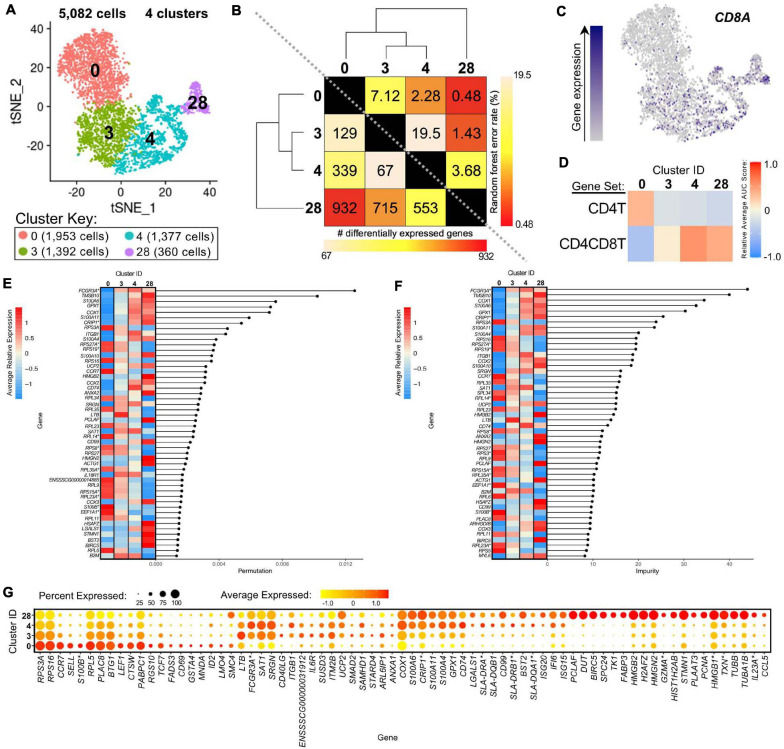
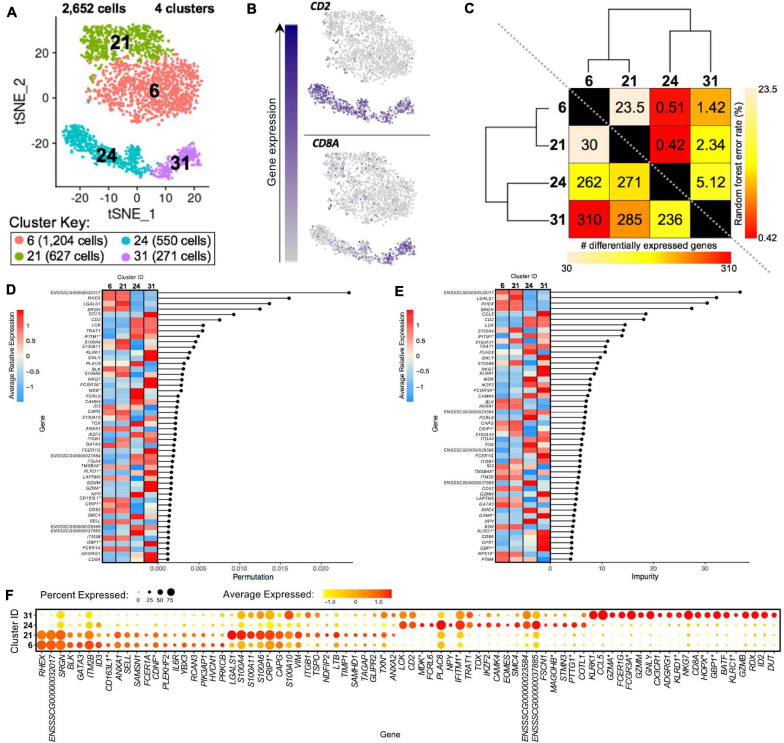
Pigs are a valuable human biomedical model and an important protein source supporting global food security. The transcriptomes of peripheral blood immune cells in pigs were defined at the bulk cell-type and single cell levels. First, eight cell types were isolated in bulk from peripheral blood mononuclear cells (PBMCs) by cell sorting, representing Myeloid, NK cells and specific populations of T and B-cells. Transcriptomes for each bulk population of cells were generated by RNA-seq with 10,974 expressed genes detected. Pairwise comparisons between cell types revealed specific expression, while enrichment analysis identified 1,885 to 3,591 significantly enriched genes across all 8 cell types. Gene Ontology analysis for the top 25% of significantly enriched genes (SEG) showed high enrichment of biological processes related to the nature of each cell type. Comparison of gene expression indicated highly significant correlations between pig cells and corresponding human PBMC bulk RNA-seq data available in Haemopedia. Second, higher resolution of distinct cell populations was obtained by single-cell RNA-sequencing (scRNA-seq) of PBMC. Seven PBMC samples were partitioned and sequenced that produced 28,810 single cell transcriptomes distributed across 36 clusters and classified into 13 general cell types including plasmacytoid dendritic cells (DC), conventional DCs, monocytes, B-cell, conventional CD4 and CD8 αβ T-cells, NK cells, and γδ T-cells. Signature gene sets from the human Haemopedia data were assessed for relative enrichment in genes expressed in pig cells and integration of pig scRNA-seq with a public human scRNA-seq dataset provided further validation for similarity between human and pig data. The sorted porcine bulk RNAseq dataset informed classification of scRNA-seq PBMC populations; specifically, an integration of the datasets showed that the pig bulk RNAseq data helped define the CD4CD8 double-positive T-cell populations in the scRNA-seq data. Overall, the data provides deep and well-validated transcriptomic data from sorted PBMC populations and the first single-cell transcriptomic data for porcine PBMCs. This resource will be invaluable for annotation of pig genes controlling immunogenetic traits as part of the porcine Functional Annotation of Animal Genomes (FAANG) project, as well as further study of, and development of new reagents for, porcine immunology.
Keywords: FAANG; bulkRNA-seq; immune cells; pig; single-cell RNA-seq; transcriptome.
Copyright © 2021 Herrera-Uribe, Wiarda, Sivasankaran, Daharsh, Liu, Byrne, Smith, Lunney, Loving and Tuggle.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Single-cell transcriptomic analysis reveals transcriptional and cell subpopulation differences between human and pig immune cells.Genes Genomics. 2024 Mar;46(3):303-322. doi: 10.1007/s13258-023-01456-9. Epub 2023 Nov 18. Genes Genomics. 2024. PMID: 37979077
-
Mapping the cellular landscape of Atlantic salmon head kidney by single cell and single nucleus transcriptomics.Fish Shellfish Immunol. 2024 Mar;146:109357. doi: 10.1016/j.fsi.2024.109357. Epub 2024 Jan 4. Fish Shellfish Immunol. 2024. PMID: 38181891
-
No detectable alloreactive transcriptional responses under standard sample preparation conditions during donor-multiplexed single-cell RNA sequencing of peripheral blood mononuclear cells.BMC Biol. 2021 Jan 20;19(1):10. doi: 10.1186/s12915-020-00941-x. BMC Biol. 2021. PMID: 33472616 Free PMC article.
-
Heterogeneity of immune cells in human atherosclerosis revealed by scRNA-Seq.Cardiovasc Res. 2021 Nov 22;117(13):2537-2543. doi: 10.1093/cvr/cvab260. Cardiovasc Res. 2021. PMID: 34343272 Free PMC article. Review.
-
Probing infectious disease by single-cell RNA sequencing: Progresses and perspectives.Comput Struct Biotechnol J. 2020;18:2962-2971. doi: 10.1016/j.csbj.2020.10.016. Epub 2020 Oct 21. Comput Struct Biotechnol J. 2020. PMID: 33106757 Free PMC article. Review.
Cited by
-
Integrated single-cell RNA-seq and DNA methylation reveal the effects of air pollution in patients with recurrent spontaneous abortion.Clin Epigenetics. 2022 Aug 23;14(1):105. doi: 10.1186/s13148-022-01327-2. Clin Epigenetics. 2022. PMID: 35999615 Free PMC article.
-
Building a FAIR data ecosystem for incorporating single-cell transcriptomics data into agricultural genome to phenome research.Front Genet. 2024 Nov 29;15:1460351. doi: 10.3389/fgene.2024.1460351. eCollection 2024. Front Genet. 2024. PMID: 39678381 Free PMC article.
-
Single-Cell RNA Sequencing Reveals the Cellular Landscape of Longissimus Dorsi in a Newborn Suhuai Pig.Int J Mol Sci. 2024 Jan 18;25(2):1204. doi: 10.3390/ijms25021204. Int J Mol Sci. 2024. PMID: 38256277 Free PMC article.
-
Spatiotemporal immune atlas of the first clinical-grade, gene-edited pig-to-human kidney xenotransplant.Res Sq [Preprint]. 2023 Jan 9:rs.3.rs-2382345. doi: 10.21203/rs.3.rs-2382345/v1. Res Sq. 2023. Update in: Nat Commun. 2024 Apr 11;15(1):3140. doi: 10.1038/s41467-024-47454-7. PMID: 36711785 Free PMC article. Updated. Preprint.
-
Single cell RNA-sequencing of feline peripheral immune cells with V(D)J repertoire and cross species analysis of T lymphocytes.Front Immunol. 2024 Nov 15;15:1438004. doi: 10.3389/fimmu.2024.1438004. eCollection 2024. Front Immunol. 2024. PMID: 39620216 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Research Materials