

A novel miR1983-TLR7-IFNβ circuit licenses NK cells to kill glioma cells, and is under the control of galectin-1
- PMID: 34249474
- PMCID: PMC8244780
- DOI: 10.1080/2162402X.2021.1939601
A novel miR1983-TLR7-IFNβ circuit licenses NK cells to kill glioma cells, and is under the control of galectin-1
Abstract
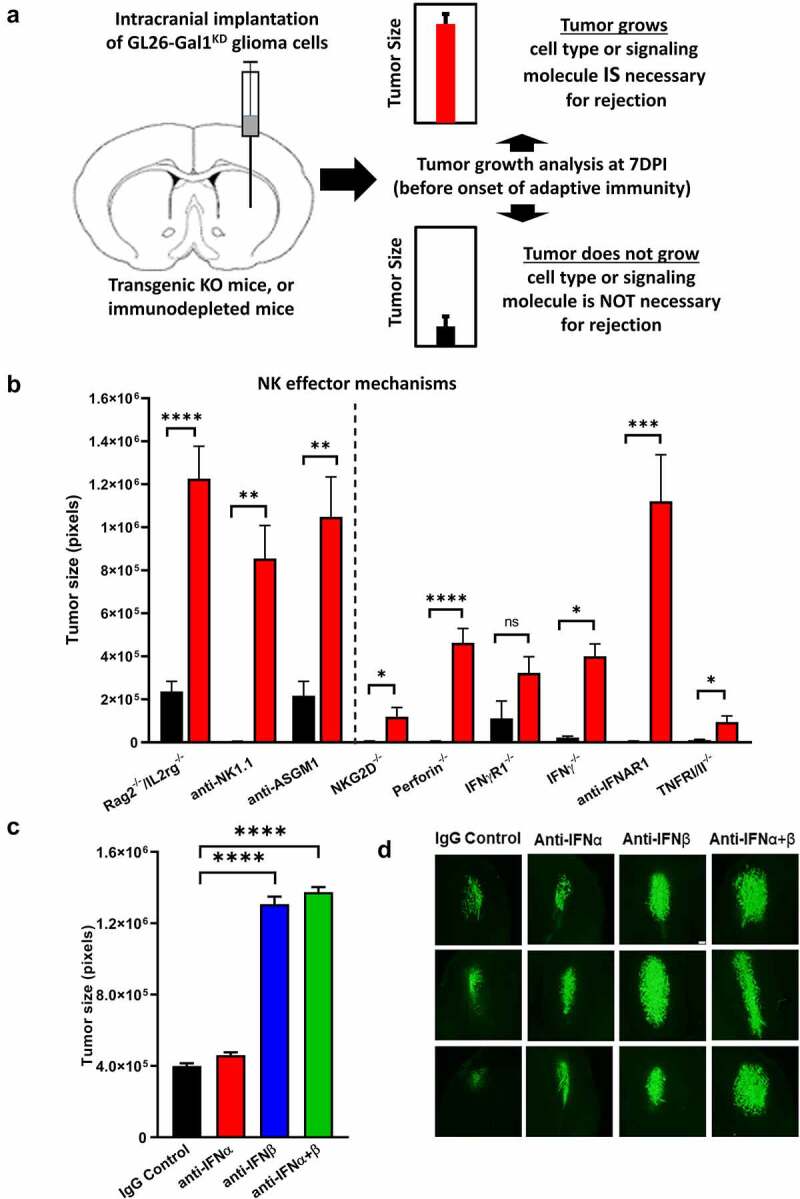
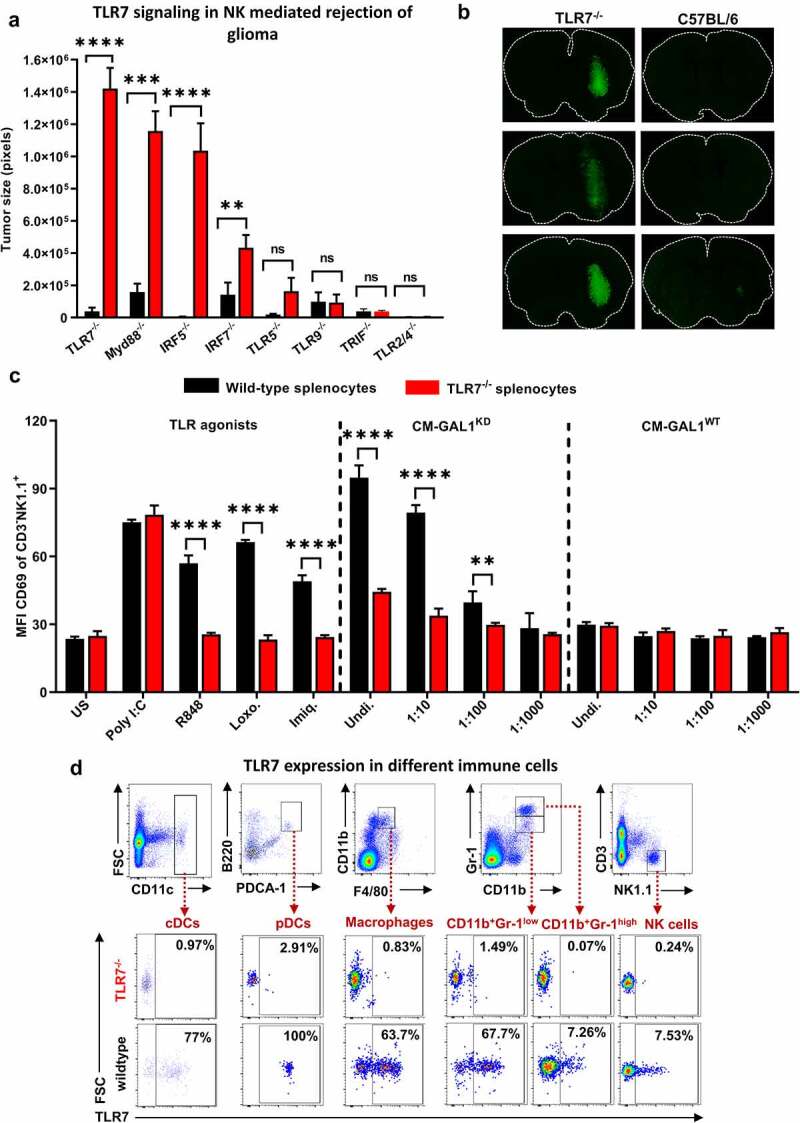
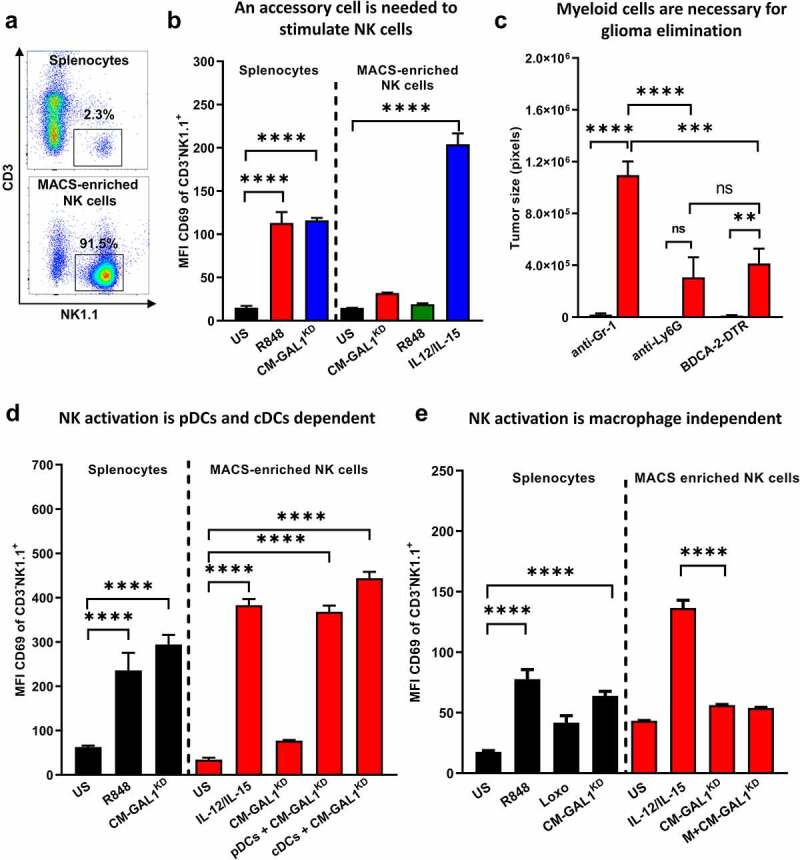
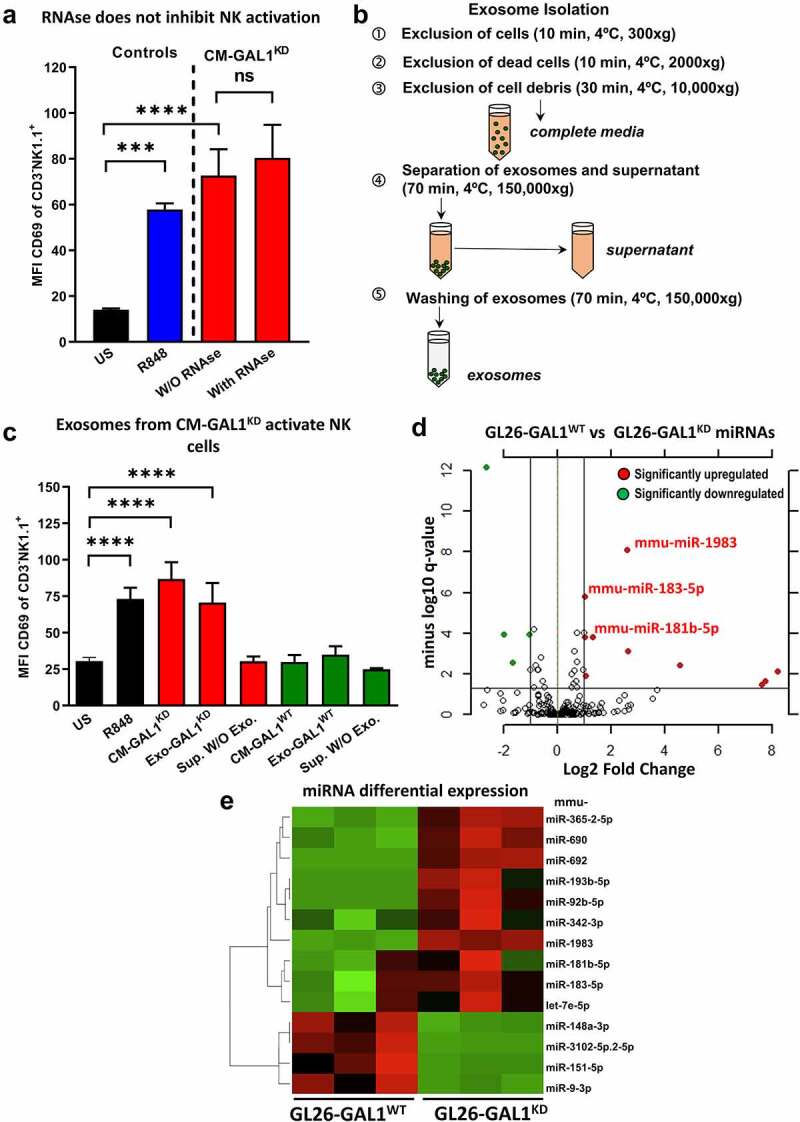
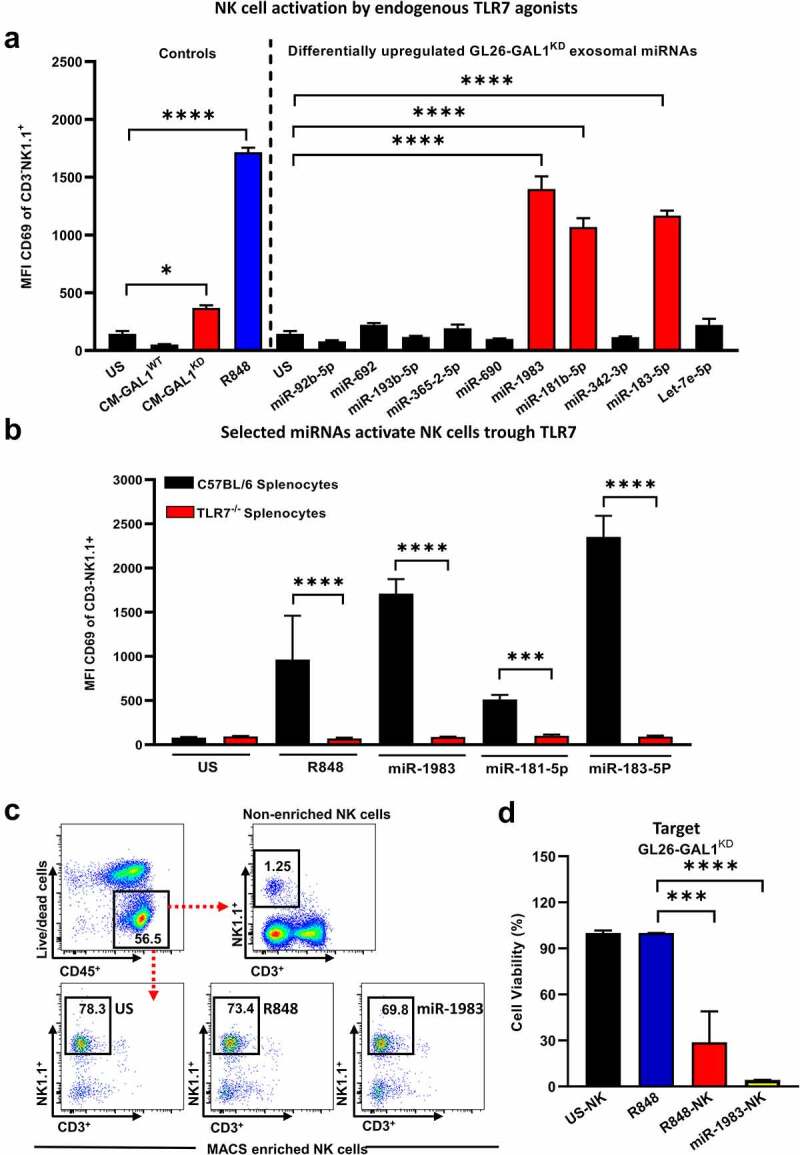
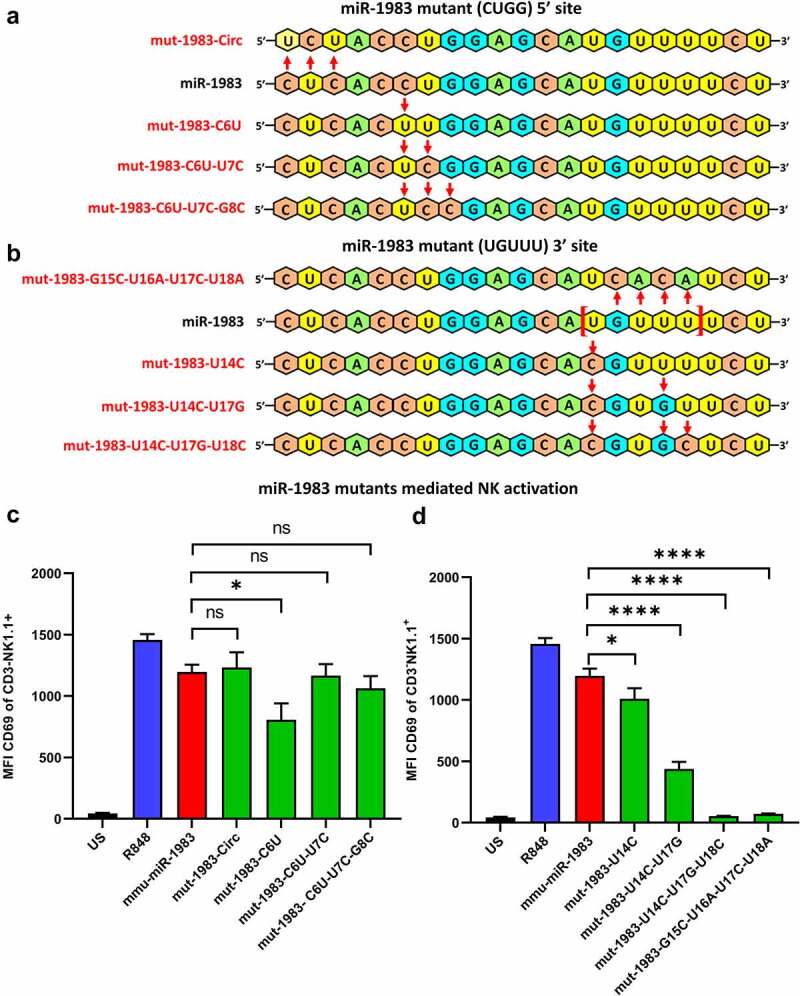
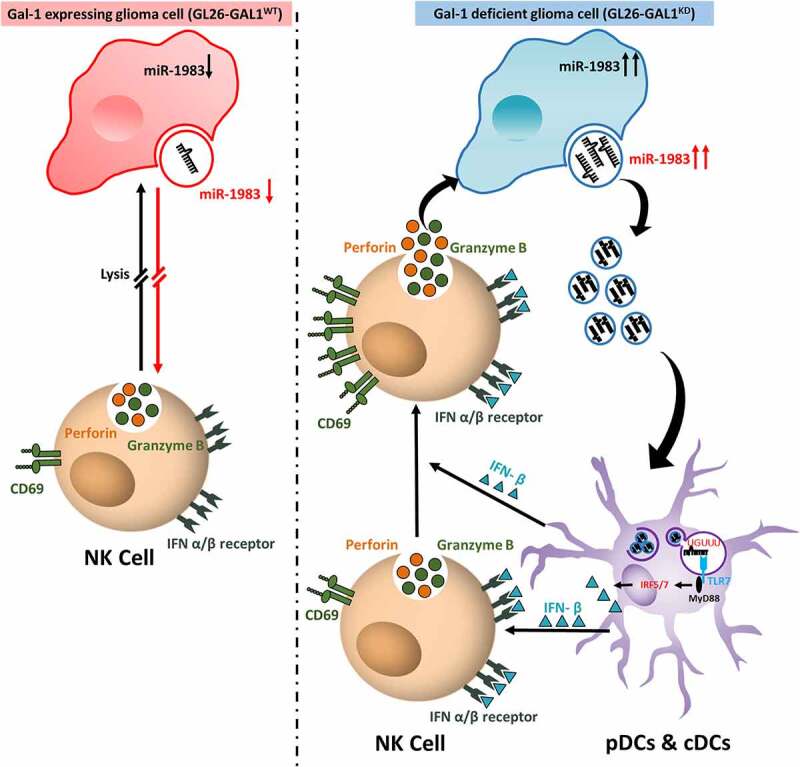
Although pharmacological stimulation of TLRs has anti-tumor effects, it has not been determined whether endogenous stimulation of TLRs can lead to tumor rejection. Herein, we demonstrate the existence of an innate anti-glioma NK-mediated circuit initiated by glioma-released miR-1983 within exosomes, and which is under the regulation of galectin-1 (Gal-1). We demonstrate that miR-1983 is an endogenous TLR7 ligand that activates TLR7 in pDCs and cDCs through a 5'-UGUUU-3' motif at its 3' end. TLR7 activation and downstream signaling through MyD88-IRF5/IRF7 stimulates secretion of IFN-β. IFN-β then stimulates NK cells resulting in the eradication of gliomas. We propose that successful immunotherapy for glioma could exploit this endogenous innate immune circuit to activate TLR7 signaling and stimulate powerful anti-glioma NK activity, at least 10-14 days before the activation of anti-tumor adaptive immunity.
Keywords: Glioblastoma; IFN-β; IRF5; IRF7; MyD88; NK cells; TLR7; galectin-1; miR-1983.
© 2021 The Author(s). Published with license by Taylor & Francis Group, LLC.
Figures
References
-
- Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, Mordoh J, Fainboim L, Podhajcer OL, Rabinovich GA. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5(3):241–251. doi:10.1016/S1535-6108(04)00024-8. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R21 NS107894/NS/NINDS NIH HHS/United States
- U01 CA224160/CA/NCI NIH HHS/United States
- R01 NS105556/NS/NINDS NIH HHS/United States
- R01 NS082311/NS/NINDS NIH HHS/United States
- R01 NS074387/NS/NINDS NIH HHS/United States
- R01 EB022563/EB/NIBIB NIH HHS/United States
- T32 CA009676/CA/NCI NIH HHS/United States
- R01 NS076991/NS/NINDS NIH HHS/United States
- UL1 TR002240/TR/NCATS NIH HHS/United States
- R37 NS094804/NS/NINDS NIH HHS/United States
- R01 NS096756/NS/NINDS NIH HHS/United States
- R21 NS091555/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials