

The Jumonji Domain-Containing Histone Demethylase Homolog 1D/lysine Demethylase 7A (JHDM1D/KDM7A) Is an Epigenetic Activator of RHOJ Transcription in Breast Cancer Cells
- PMID: 34249916
- PMCID: PMC8262595
- DOI: 10.3389/fcell.2021.664375
The Jumonji Domain-Containing Histone Demethylase Homolog 1D/lysine Demethylase 7A (JHDM1D/KDM7A) Is an Epigenetic Activator of RHOJ Transcription in Breast Cancer Cells
Erratum in
-
Corrigendum: The Jumonji Domain-Containing Histone Demethylase Homolog 1D/lysine Demethylase 7A (JHDM1D/KDM7A) Is an Epigenetic Activator of RHOJ Transcription in Breast Cancer Cells.Front Cell Dev Biol. 2021 Jul 19;9:729416. doi: 10.3389/fcell.2021.729416. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34395453 Free PMC article.
Abstract
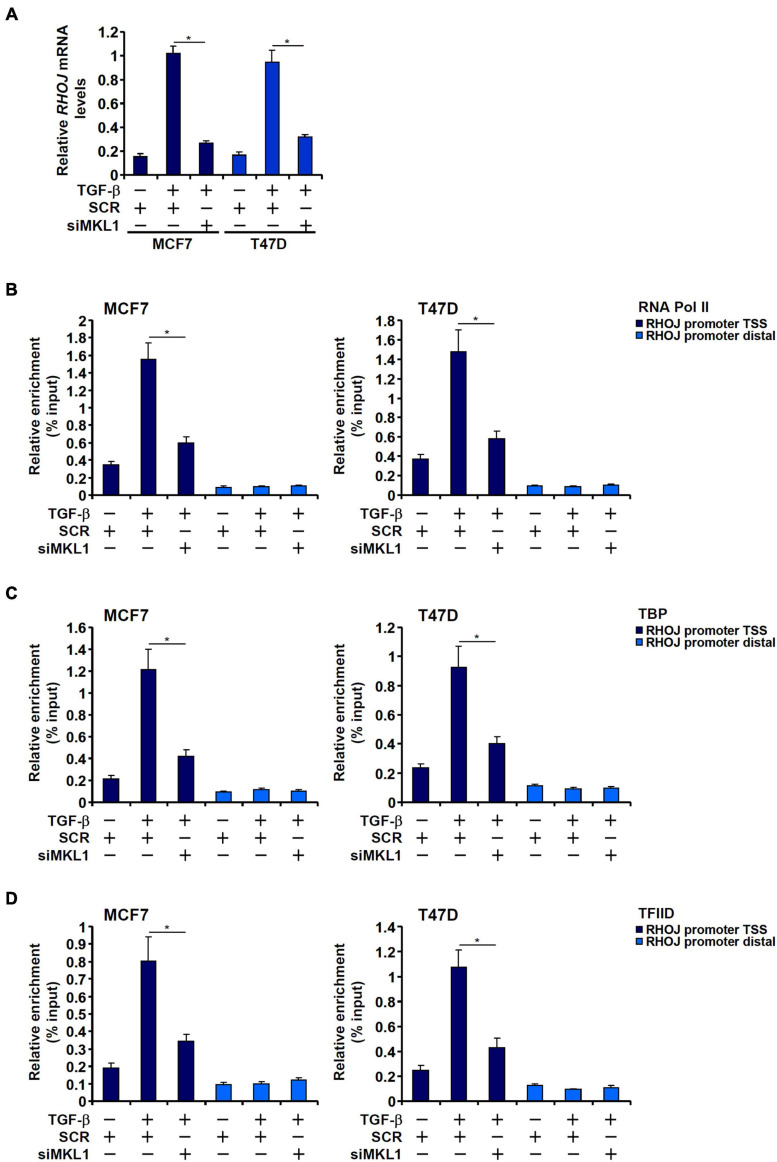
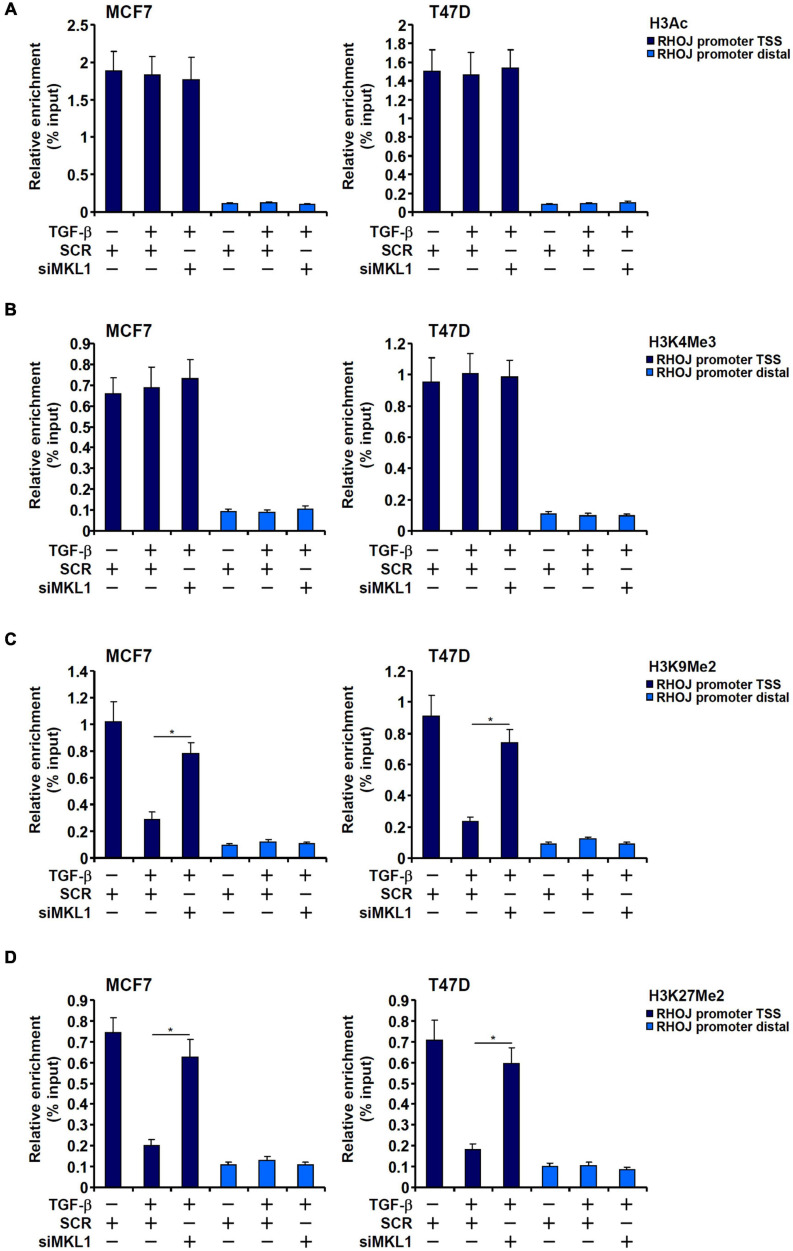
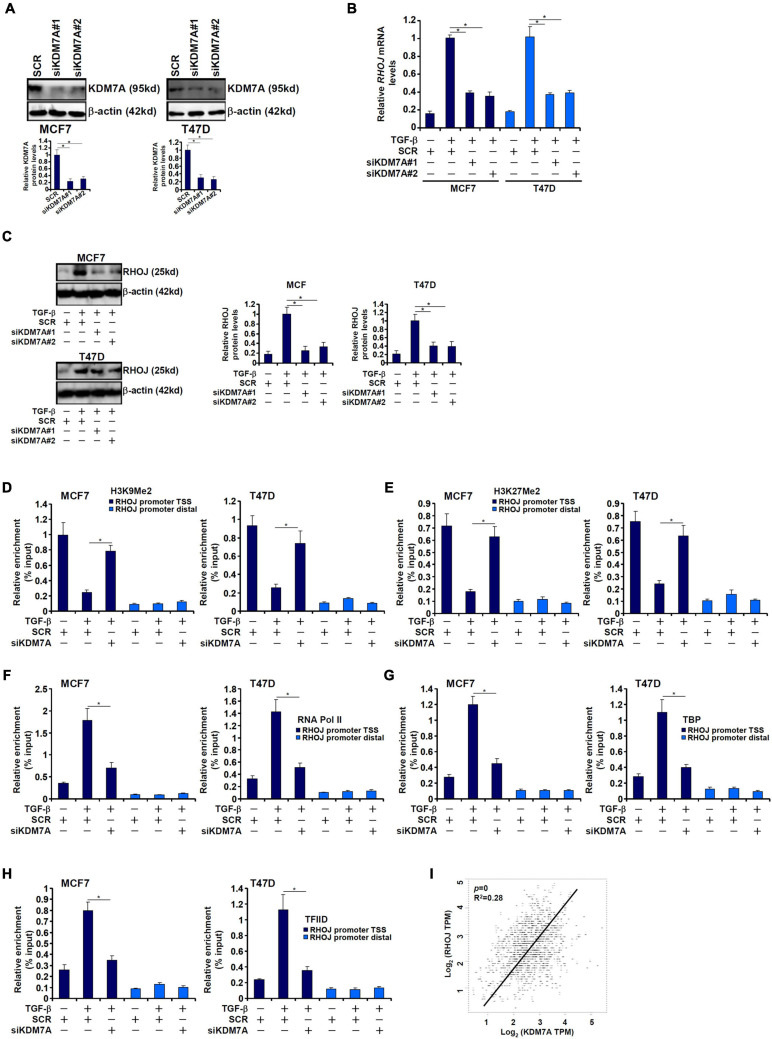
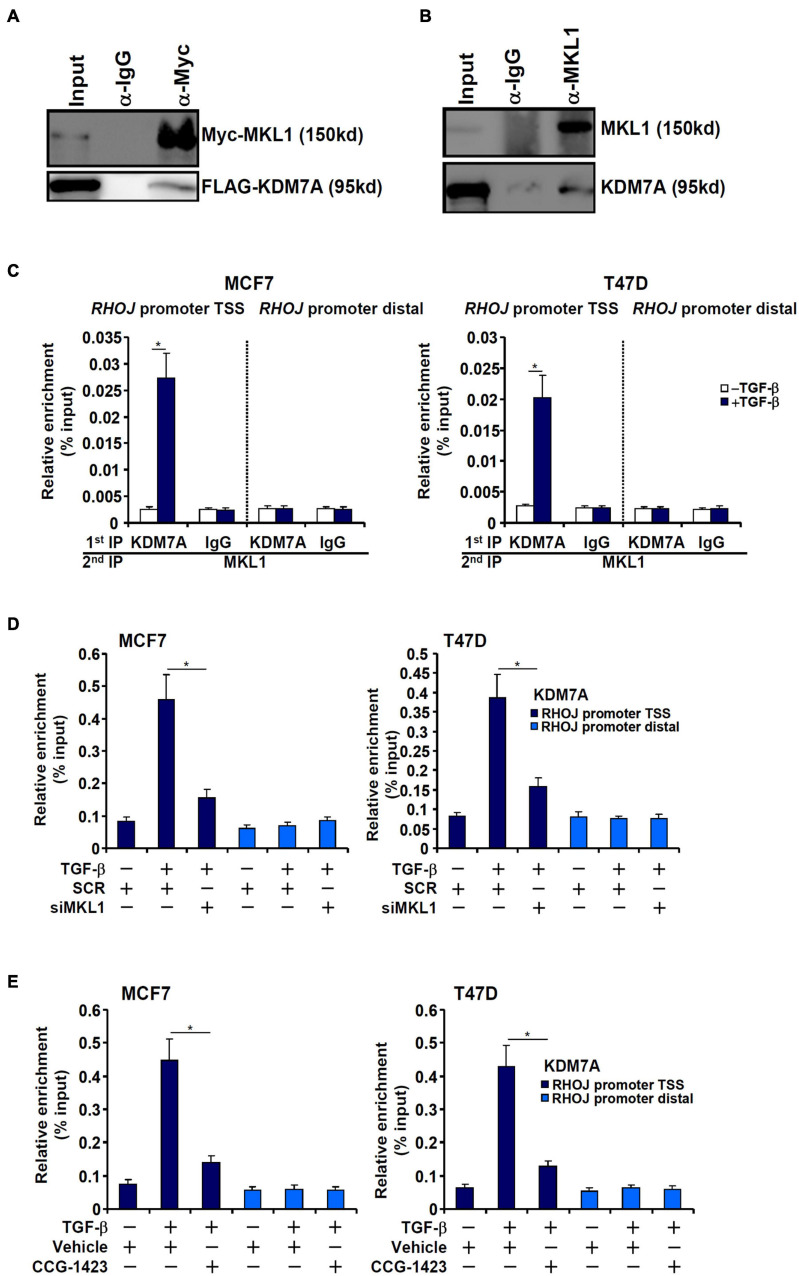
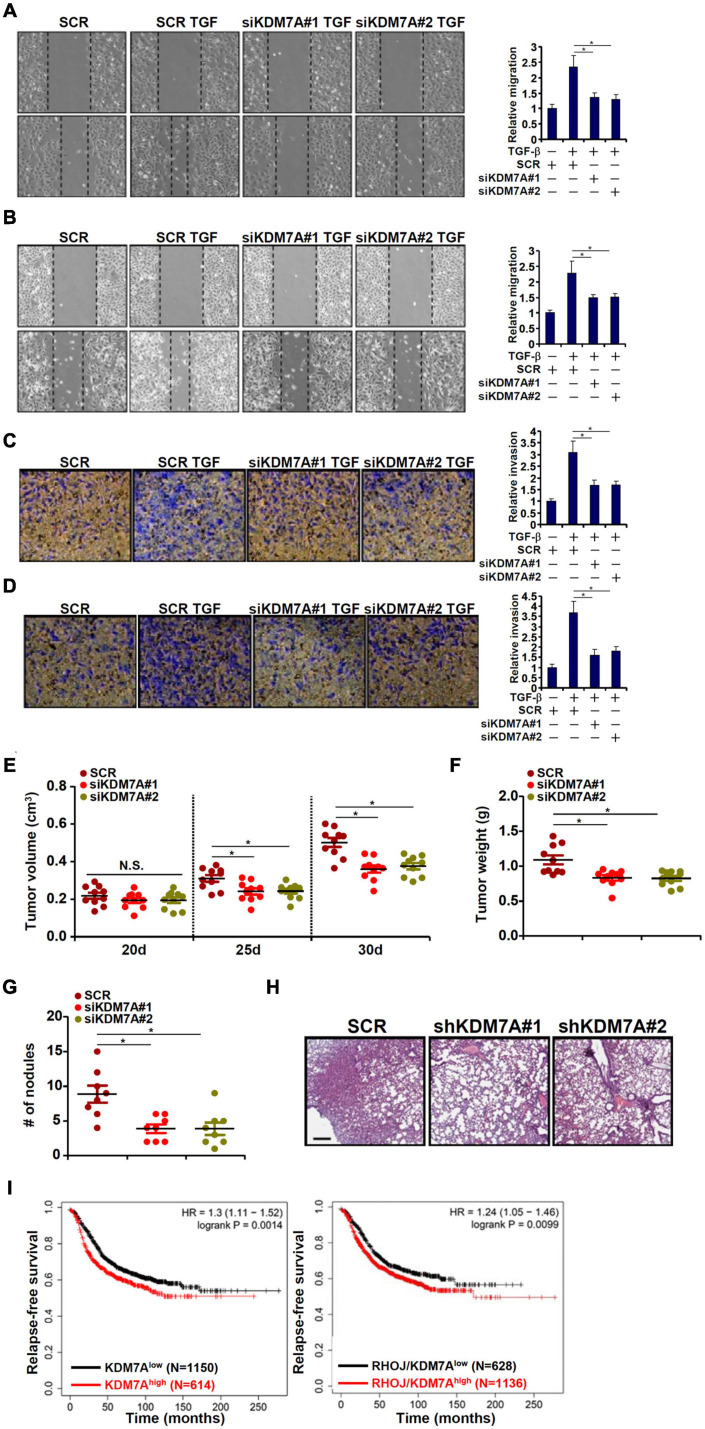
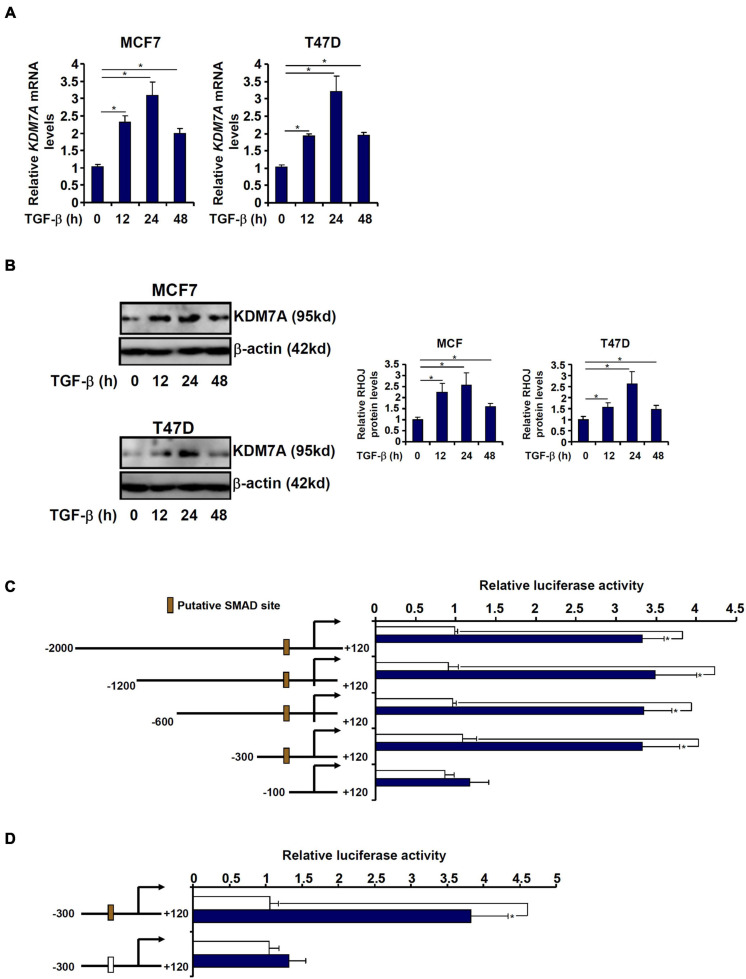
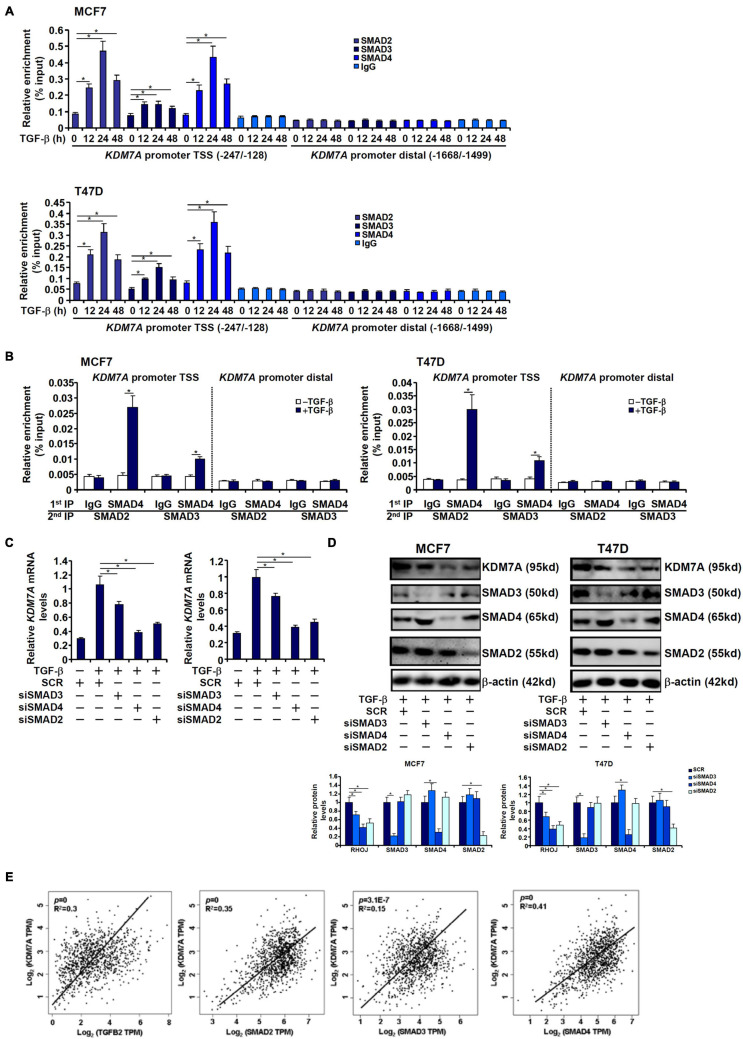
The small GTPase RHOJ is a key regulator of breast cancer metastasis by promoting cell migration and invasion. The prometastatic stimulus TGF-β activates RHOJ transcription via megakaryocytic leukemia 1 (MKL1). The underlying epigenetic mechanism is not clear. Here, we report that MKL1 deficiency led to disrupted assembly of the RNA polymerase II preinitiation complex on the RHOJ promoter in breast cancer cells. This could be partially explained by histone H3K9/H3K27 methylation status. Further analysis confirmed that the H3K9/H3K27 dual demethylase JHDM1D/KDM7A was essential for TGF-β-induced RHOJ transcription in breast cancer cells. MKL1 interacted with and recruited KDM7A to the RHOJ promoter to cooperatively activate RHOJ transcription. KDM7A knockdown attenuated migration and invasion of breast cancer cells in vitro and mitigated the growth and metastasis of breast cancer cells in nude mice. KDM7A expression level, either singularly or in combination with that of RHOJ, could be used to predict prognosis in breast cancer patients. Of interest, KDM7A appeared to be a direct transcriptional target of TGF-β signaling. A SMAD2/SMAD4 complex bound to the KDM7A promoter and mediated TGF-β-induced KDM7A transcription. In conclusion, our data unveil a novel epigenetic mechanism whereby TGF-β regulates the transcription of the prometastatic small GTPase RHOJ. Screening for small-molecule inhibitors of KDM7A may yield effective therapeutic solutions to treat malignant breast cancers.
Keywords: breast cancer cell; epigenetics; histone demethylase; histone methylation; transcriptional regulation.
Copyright © 2021 Zhang, Chen, Zhu, Zhang, Yuan, Zhang and Xu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Corrigendum: The Jumonji Domain-Containing Histone Demethylase Homolog 1D/lysine Demethylase 7A (JHDM1D/KDM7A) Is an Epigenetic Activator of RHOJ Transcription in Breast Cancer Cells.Front Cell Dev Biol. 2021 Jul 19;9:729416. doi: 10.3389/fcell.2021.729416. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34395453 Free PMC article.
-
MKL1 Mediates TGF-β Induced RhoJ Transcription to Promote Breast Cancer Cell Migration and Invasion.Front Cell Dev Biol. 2020 Aug 25;8:832. doi: 10.3389/fcell.2020.00832. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32984327 Free PMC article.
-
Histone Demethylase KDM7A Regulates Androgen Receptor Activity, and Its Chemical Inhibitor TC-E 5002 Overcomes Cisplatin-Resistance in Bladder Cancer Cells.Int J Mol Sci. 2020 Aug 6;21(16):5658. doi: 10.3390/ijms21165658. Int J Mol Sci. 2020. PMID: 32781788 Free PMC article.
-
Lysine-specific demethylase 7A (KDM7A): A potential target for disease therapy.Biochem Pharmacol. 2023 Oct;216:115799. doi: 10.1016/j.bcp.2023.115799. Epub 2023 Sep 9. Biochem Pharmacol. 2023. PMID: 37696455 Review.
-
Kdm7a expression is spatiotemporally regulated in developing Xenopus laevis embryos, and its overexpression influences late retinal development.Dev Dyn. 2024 May;253(5):508-518. doi: 10.1002/dvdy.670. Epub 2023 Nov 1. Dev Dyn. 2024. PMID: 37909656 Review.
Cited by
-
Current advances and future directions in targeting histone demethylases for cancer therapy.Mol Cells. 2025 Mar;48(3):100192. doi: 10.1016/j.mocell.2025.100192. Epub 2025 Feb 10. Mol Cells. 2025. PMID: 39938867 Free PMC article. Review.
-
Nuclear localization of BRCA1-associated protein 1 is important in suppressing hepatocellular carcinoma metastasis via CTCF and NRF1/OGT axis.Cell Death Dis. 2025 Feb 21;16(1):123. doi: 10.1038/s41419-025-07451-0. Cell Death Dis. 2025. PMID: 39984455 Free PMC article.
-
HES5-mediated repression of LIGHT transcription may contribute to apoptosis in hepatocytes.Cell Death Discov. 2021 Oct 23;7(1):308. doi: 10.1038/s41420-021-00707-6. Cell Death Discov. 2021. PMID: 34689159 Free PMC article.
-
Dual Regulation of Tank Binding Kinase 1 by BRG1 in Hepatocytes Contributes to Reactive Oxygen Species Production.Front Cell Dev Biol. 2021 Oct 1;9:745985. doi: 10.3389/fcell.2021.745985. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34660604 Free PMC article.
-
An E2F5-TFDP1-BRG1 Complex Mediates Transcriptional Activation of MYCN in Hepatocytes.Front Cell Dev Biol. 2021 Oct 22;9:742319. doi: 10.3389/fcell.2021.742319. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34746136 Free PMC article.
References
-
- Brower V. (2011). Epigenetics: unravelling the cancer code. Nature 471 S12–S13. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous