

Profiling variable-number tandem repeat variation across populations using repeat-pangenome graphs
- PMID: 34253730
- PMCID: PMC8275641
- DOI: 10.1038/s41467-021-24378-0
Profiling variable-number tandem repeat variation across populations using repeat-pangenome graphs
Abstract
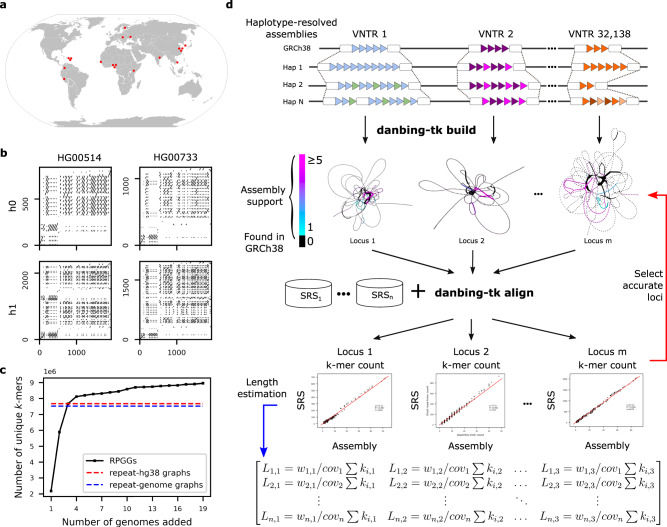
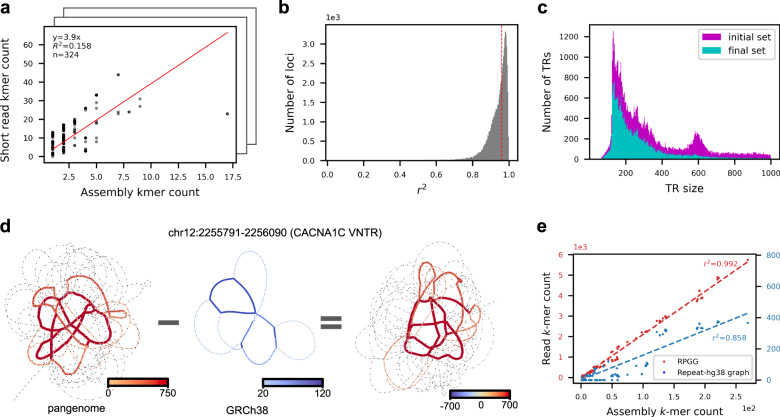
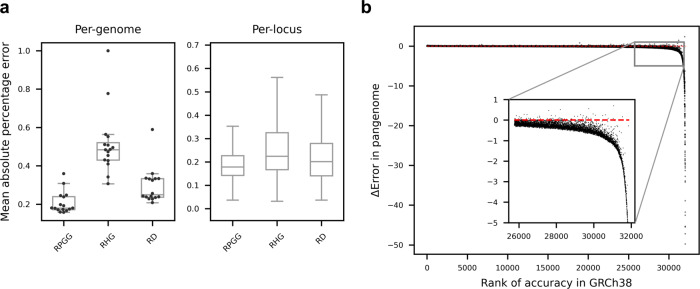
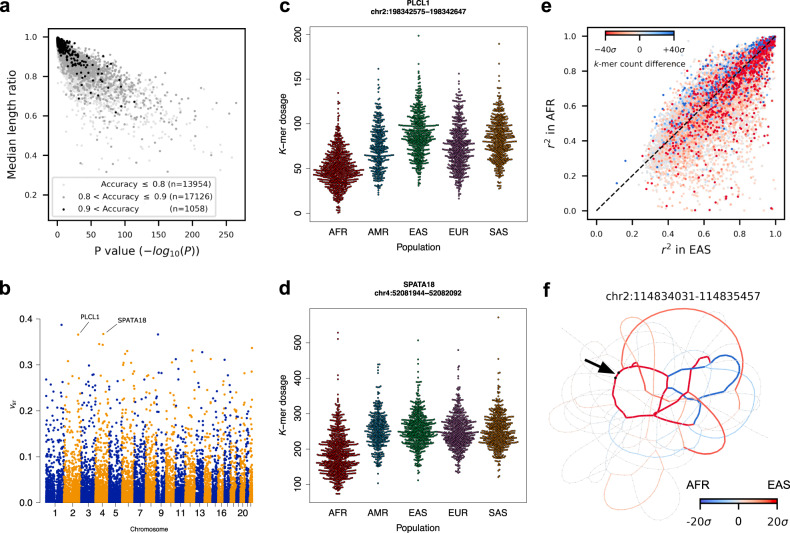
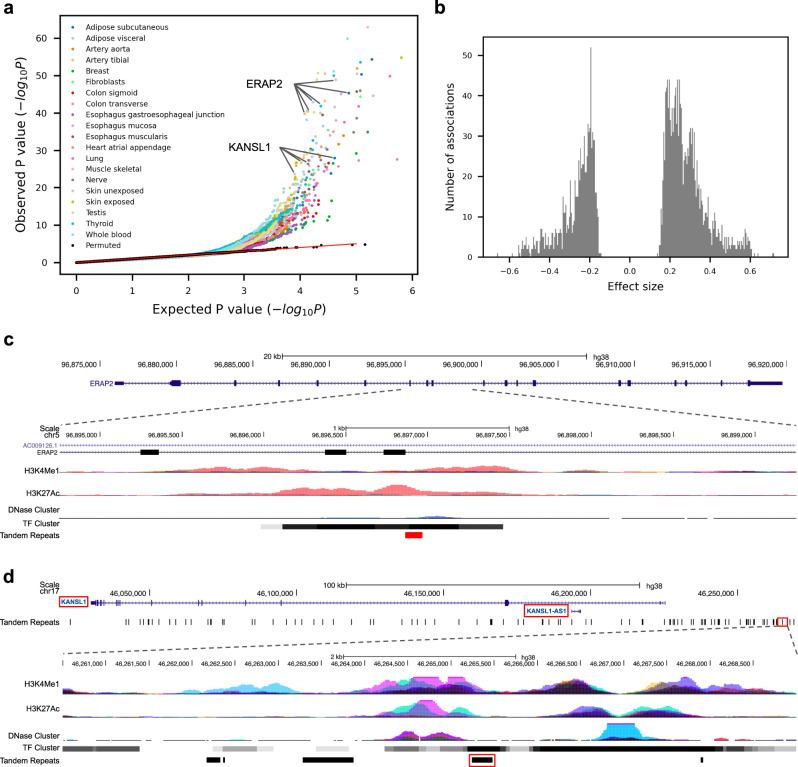
Variable number tandem repeats (VNTRs) are composed of consecutive repetitive DNA with hypervariable repeat count and composition. They include protein coding sequences and associations with clinical disorders. It has been difficult to incorporate VNTR analysis in disease studies that use short-read sequencing because the traditional approach of mapping to the human reference is less effective for repetitive and divergent sequences. In this work, we solve VNTR mapping for short reads with a repeat-pangenome graph (RPGG), a data structure that encodes both the population diversity and repeat structure of VNTR loci from multiple haplotype-resolved assemblies. We develop software to build a RPGG, and use the RPGG to estimate VNTR composition with short reads. We use this to discover VNTRs with length stratified by continental population, and expression quantitative trait loci, indicating that RPGG analysis of VNTRs will be critical for future studies of diversity and disease.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
