

Chemotherapy-induced transposable elements activate MDA5 to enhance haematopoietic regeneration
- PMID: 34253898
- PMCID: PMC8492473
- DOI: 10.1038/s41556-021-00707-9
Chemotherapy-induced transposable elements activate MDA5 to enhance haematopoietic regeneration
Erratum in
-
Publisher Correction: Chemotherapy-induced transposable elements activate MDA5 to enhance haematopoietic regeneration.Nat Cell Biol. 2021 Dec;23(12):1338. doi: 10.1038/s41556-021-00785-9. Nat Cell Biol. 2021. PMID: 34611323 Free PMC article. No abstract available.
Abstract
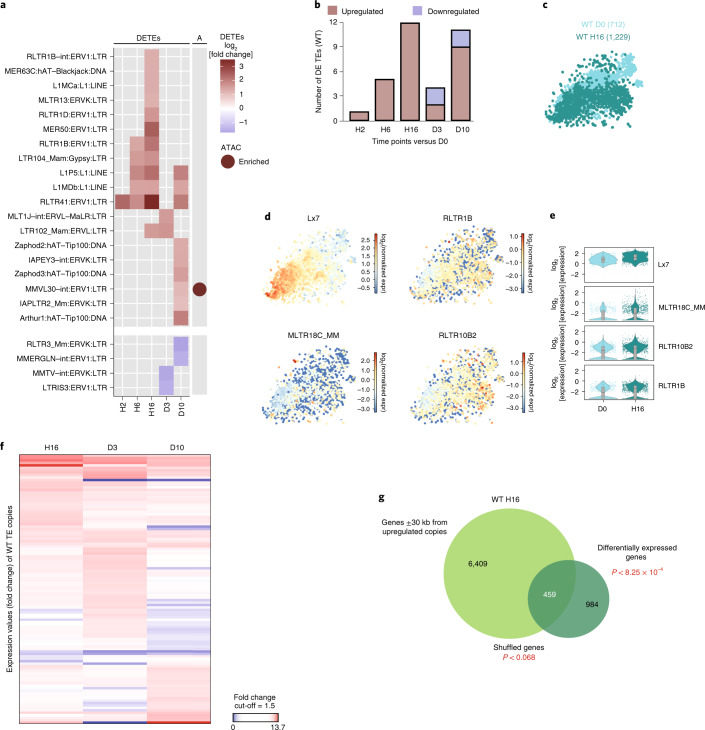
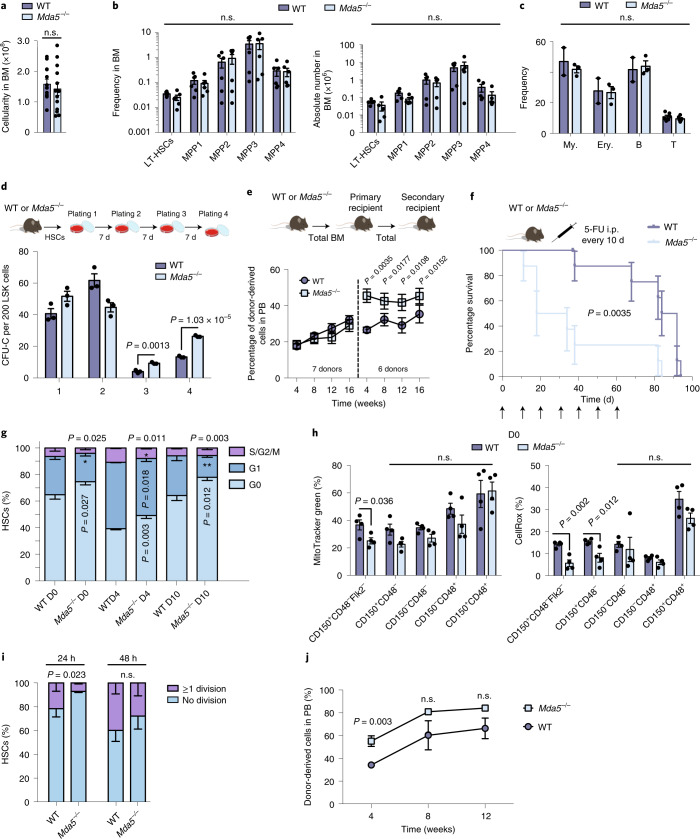
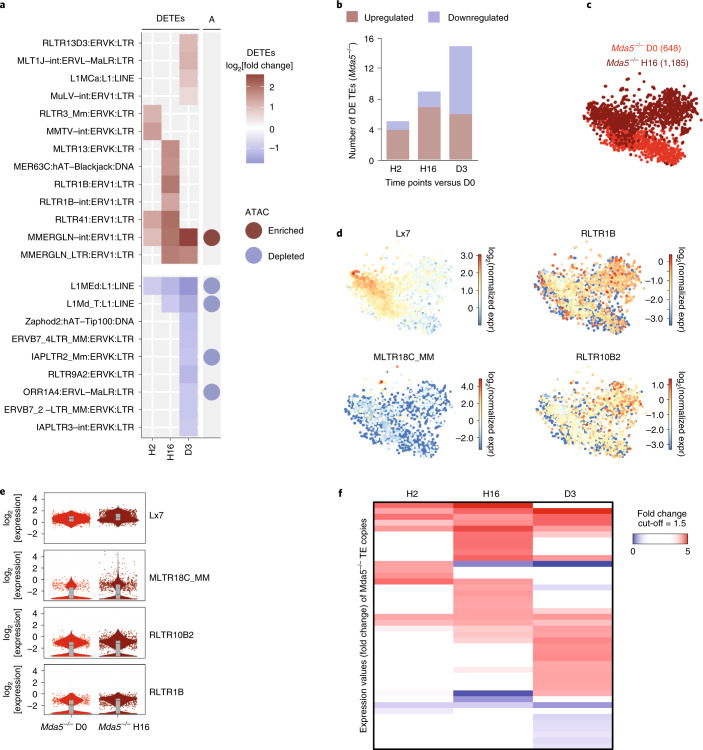
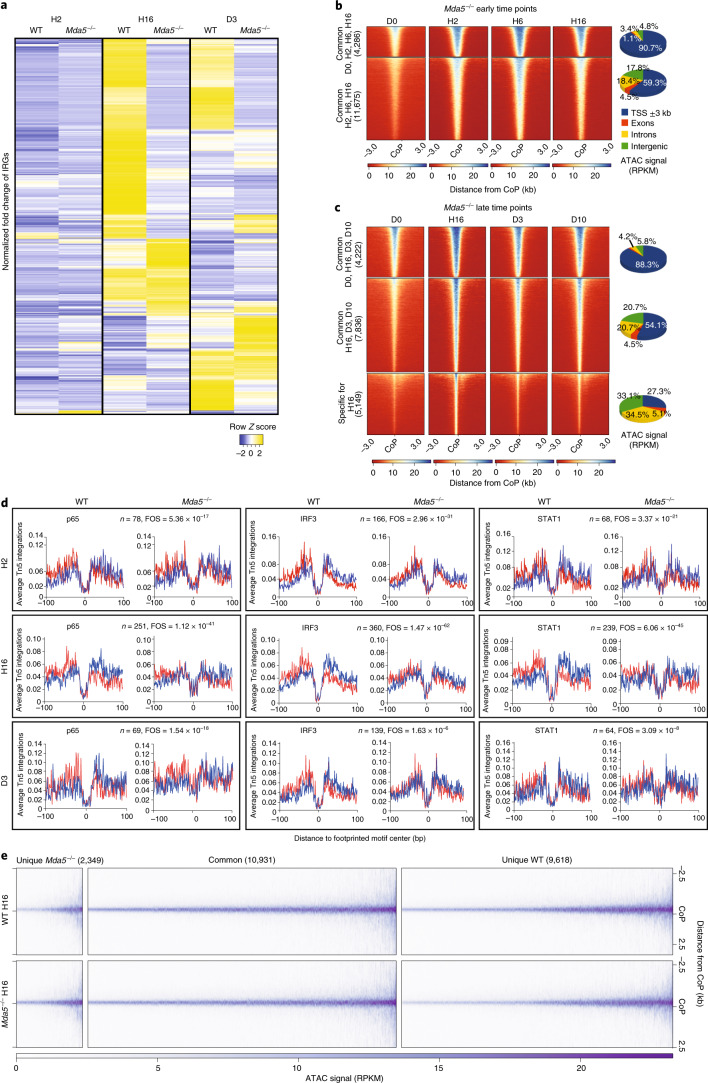
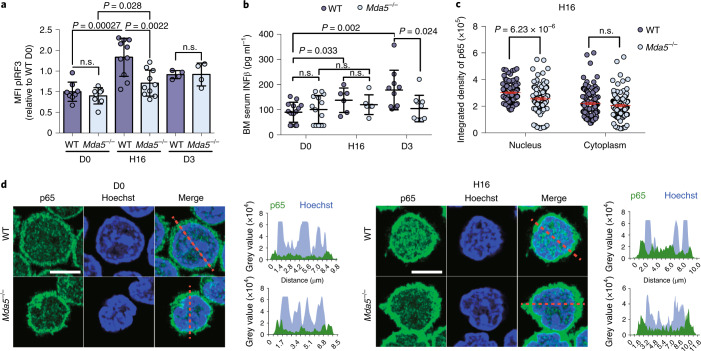
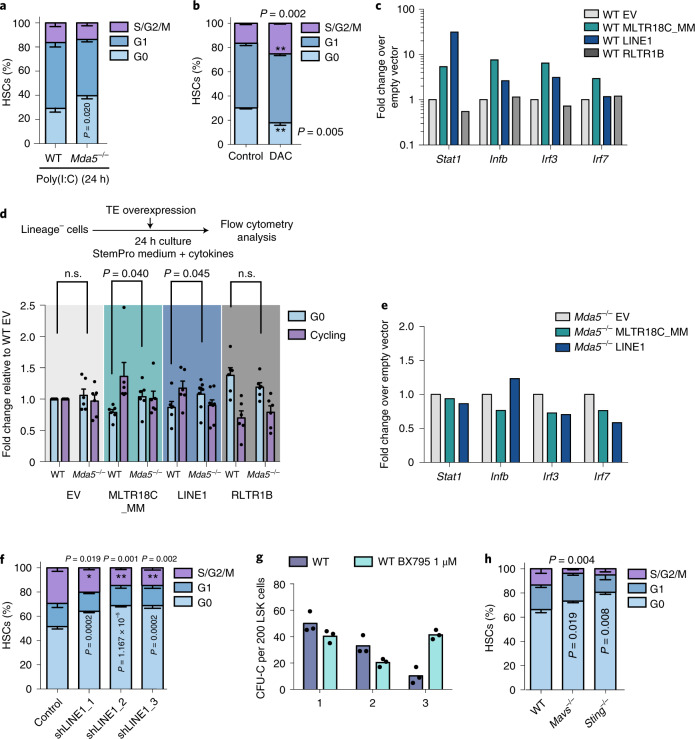
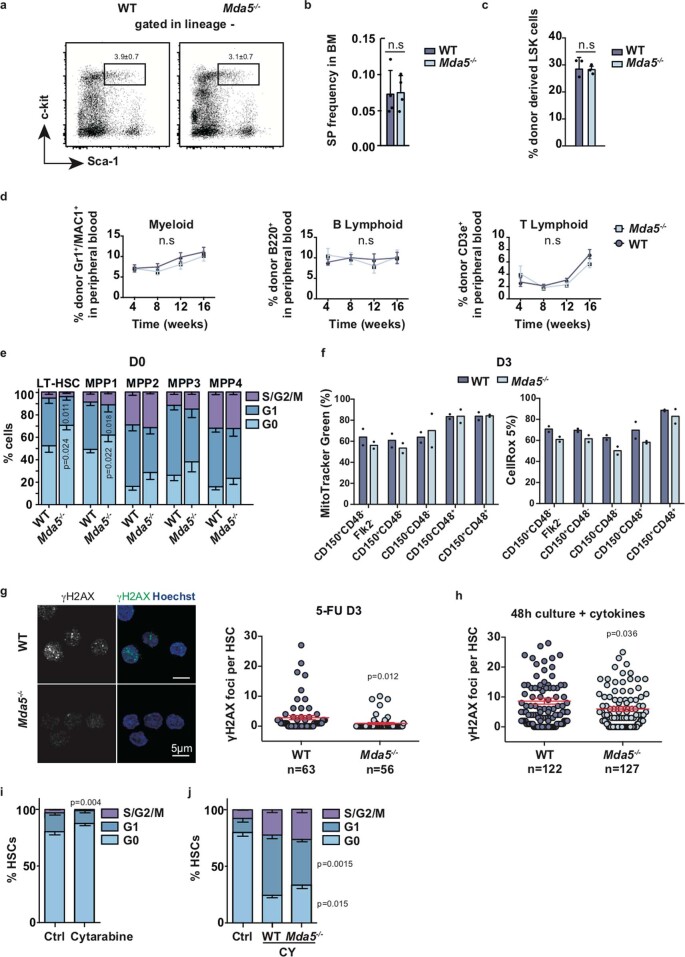
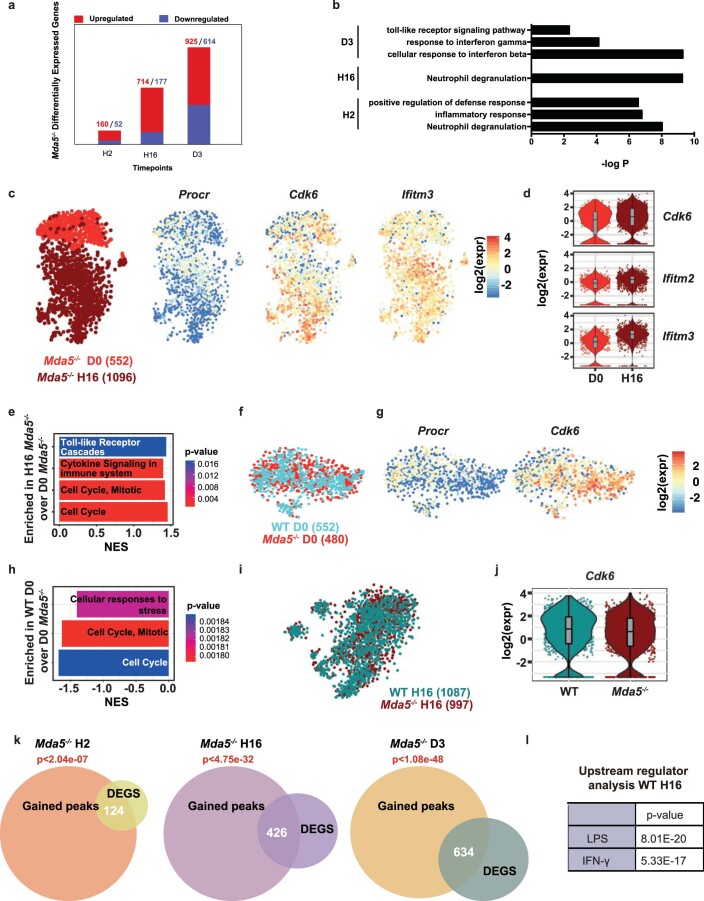
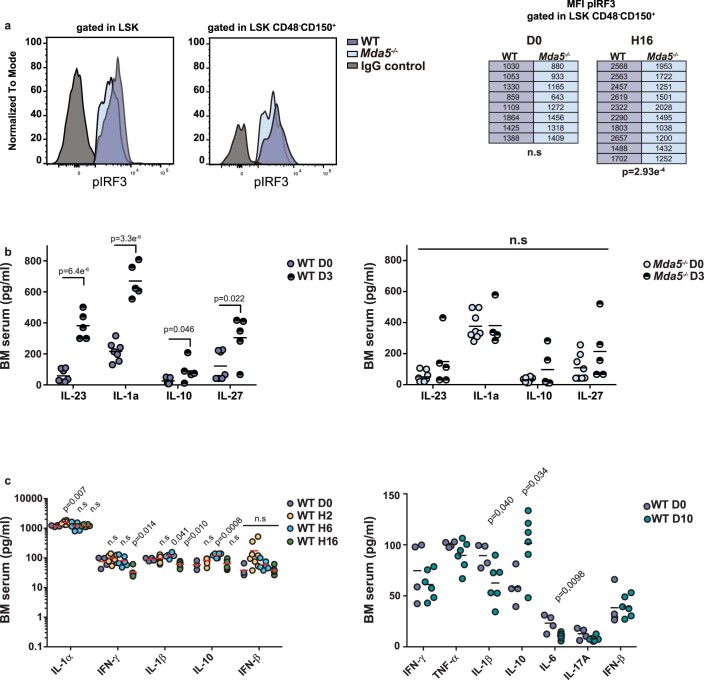
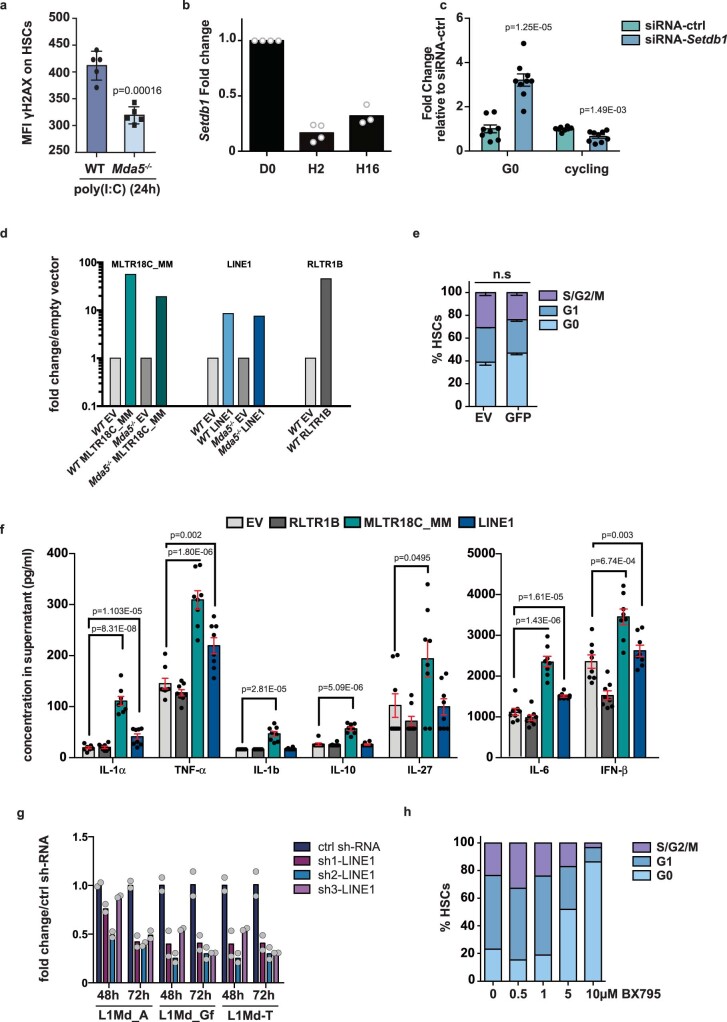
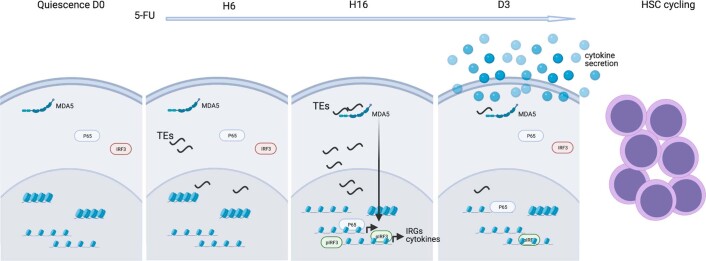
Haematopoietic stem cells (HSCs) are normally quiescent, but have evolved mechanisms to respond to stress. Here, we evaluate haematopoietic regeneration induced by chemotherapy. We detect robust chromatin reorganization followed by increased transcription of transposable elements (TEs) during early recovery. TE transcripts bind to and activate the innate immune receptor melanoma differentiation-associated protein 5 (MDA5) that generates an inflammatory response that is necessary for HSCs to exit quiescence. HSCs that lack MDA5 exhibit an impaired inflammatory response after chemotherapy and retain their quiescence, with consequent better long-term repopulation capacity. We show that the overexpression of ERV and LINE superfamily TE copies in wild-type HSCs, but not in Mda5-/- HSCs, results in their cycling. By contrast, after knockdown of LINE1 family copies, HSCs retain their quiescence. Our results show that TE transcripts act as ligands that activate MDA5 during haematopoietic regeneration, thereby enabling HSCs to mount an inflammatory response necessary for their exit from quiescence.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010;10:201–209. - PubMed
-
- He S, Nakada D, Morrison SJ. Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol. 2009;25:377–406. - PubMed
-
- Essers MA, et al. IFNα activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
