

Vascular Mechanobiology: Homeostasis, Adaptation, and Disease
- PMID: 34255994
- PMCID: PMC8719655
- DOI: 10.1146/annurev-bioeng-092419-060810
Vascular Mechanobiology: Homeostasis, Adaptation, and Disease
Abstract
Cells of the vascular wall are exquisitely sensitive to changes in their mechanical environment. In healthy vessels, mechanical forces regulate signaling and gene expression to direct the remodeling needed for the vessel wall to maintain optimal function. Major diseases of arteries involve maladaptive remodeling with compromised or lost homeostatic mechanisms. Whereas homeostasis invokes negative feedback loops at multiple scales to mediate mechanobiological stability, disease progression often occurs via positive feedback that generates mechanobiological instabilities. In this review, we focus on the cell biology, wall mechanics, and regulatory pathways associated with arterial health and how changes in these processes lead to disease. We discuss how positive feedback loops arise via biomechanical and biochemical means. We conclude that inflammation plays a central role in overriding homeostatic pathways and suggest future directions for addressing therapeutic needs.
Keywords: aneurysms; atherosclerosis; feedback; fibrosis; hypertension; mechanotransduction.
Figures
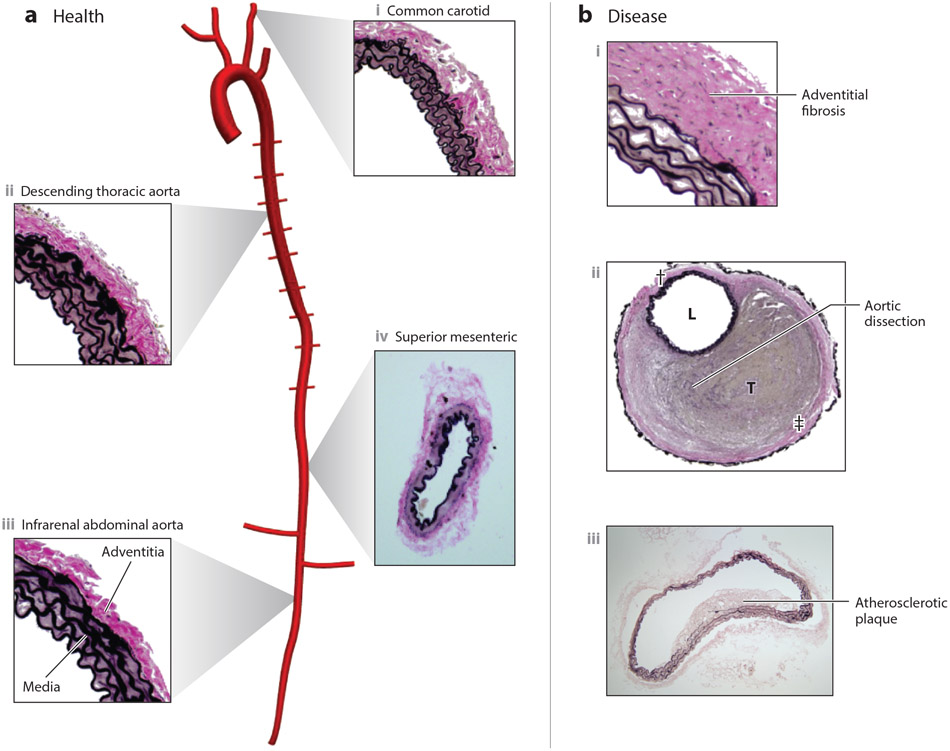
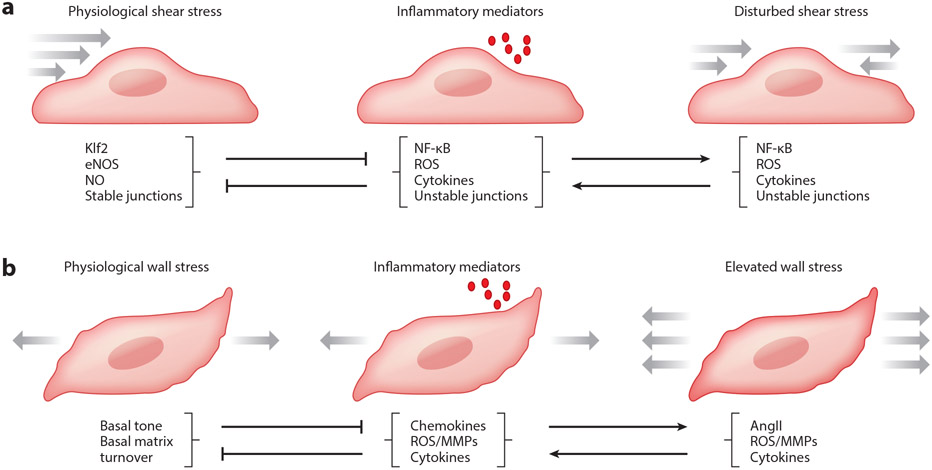
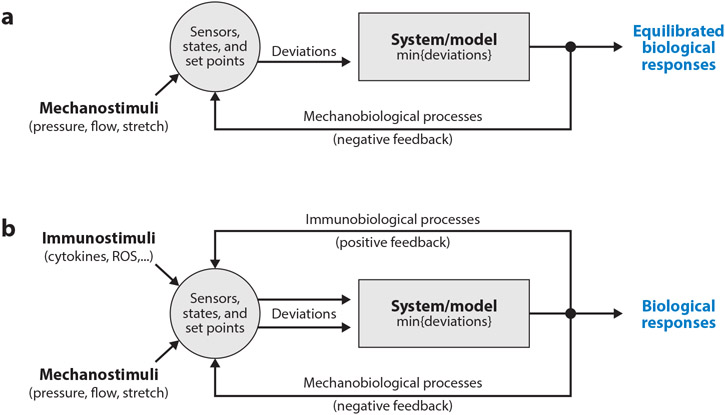
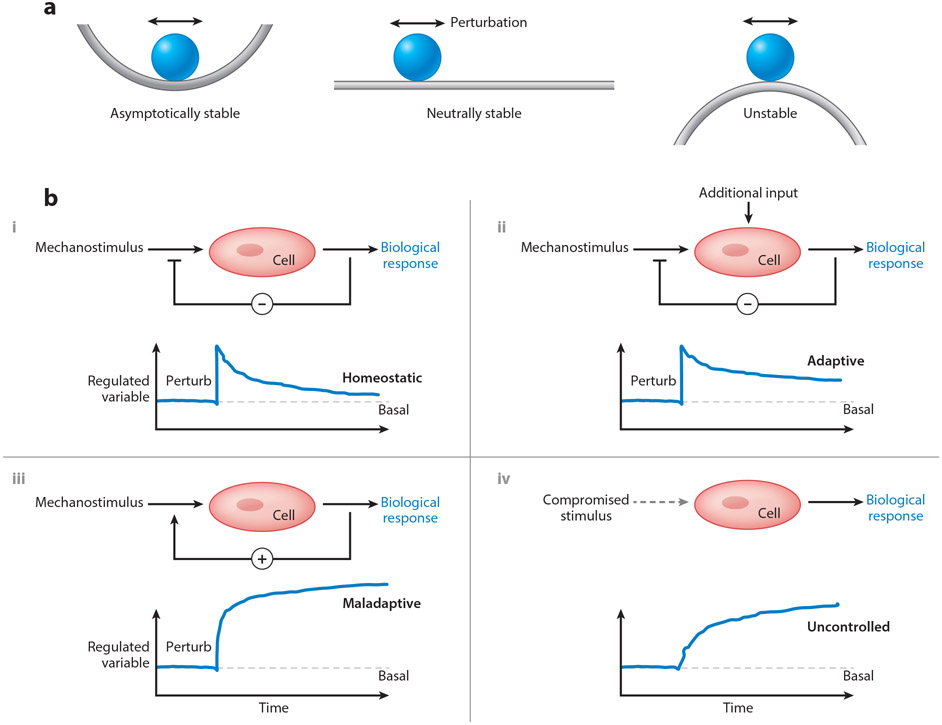
References
-
- Dajnowiec D, Langille BL. 2007. Arterial adaptations to chronic changes in haemodynamic function: coupling vasomotor tone to structural remodelling. Clin Sci (Lond) 113:15–23 - PubMed
-
- Bikfalvi A 2006. Angiogenesis: health and disease. Ann Oncol 17 Suppl 10:x65–70 - PubMed
-
- Safar ME. 2010. Arterial aging--hemodynamic changes and therapeutic options. Nat Rev Cardiol 7:442–9 - PubMed
-
- Humphrey JD. 2002. Cardiovascular Solid Mechanics: Cells, Tissues, and Organs. Springer-Verlag; New York. XVI, 758 pp.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
