

The role of interspecies recombination in the evolution of antibiotic-resistant pneumococci
- PMID: 34259624
- PMCID: PMC8321556
- DOI: 10.7554/eLife.67113
The role of interspecies recombination in the evolution of antibiotic-resistant pneumococci
Abstract
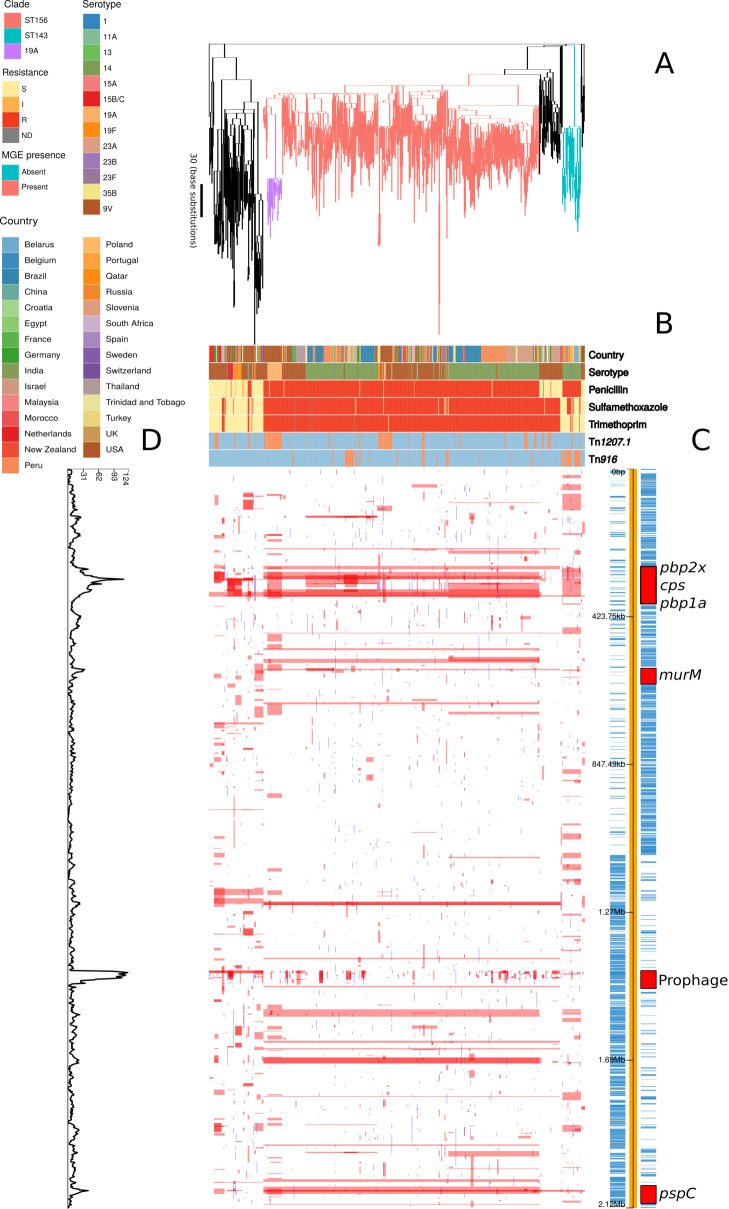
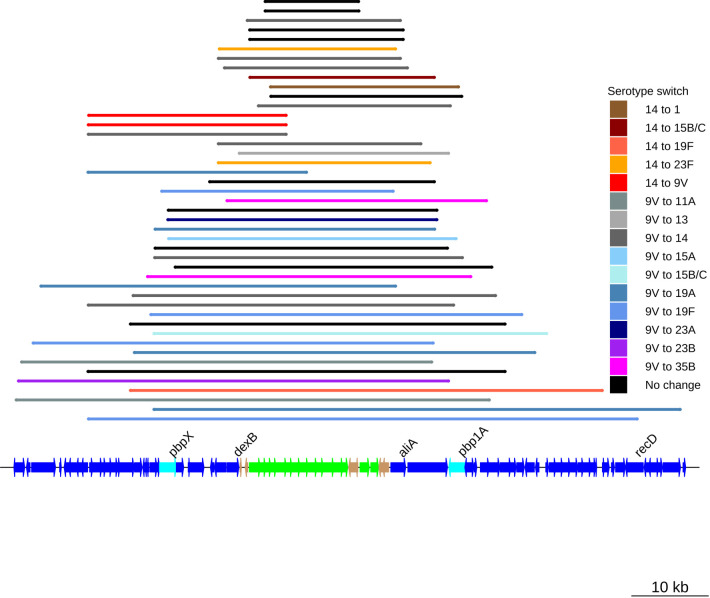
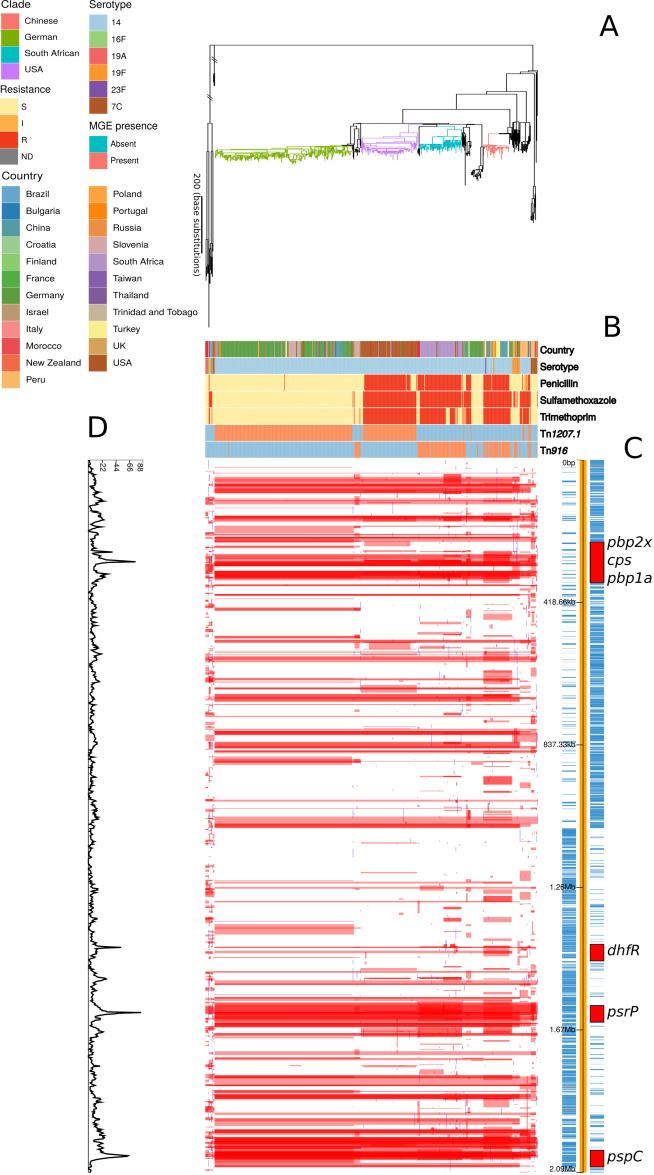
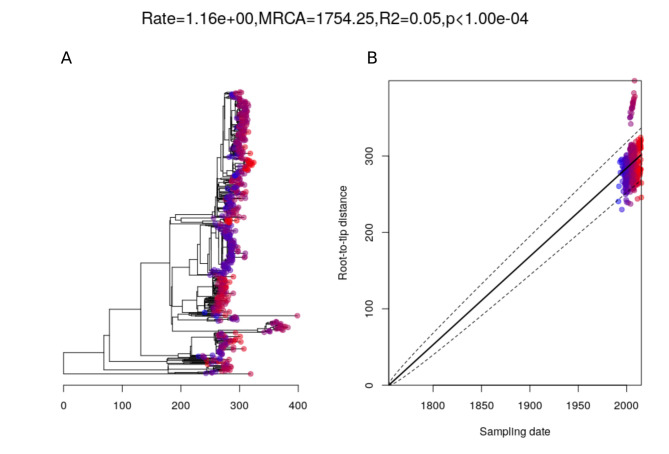
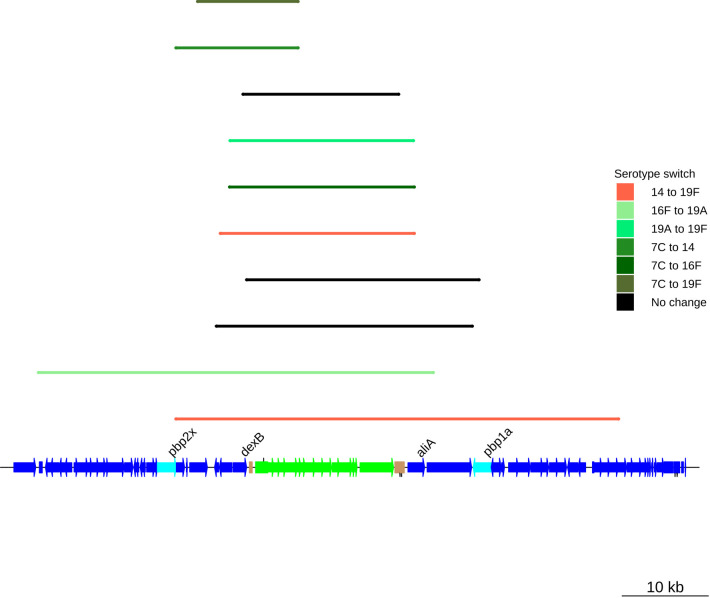
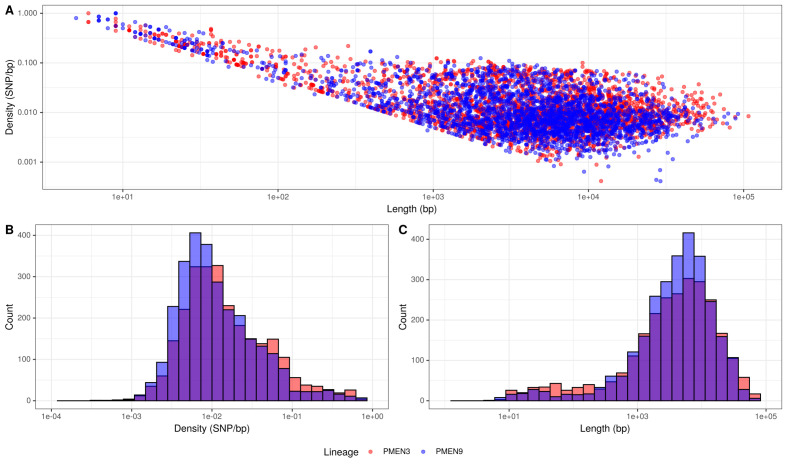
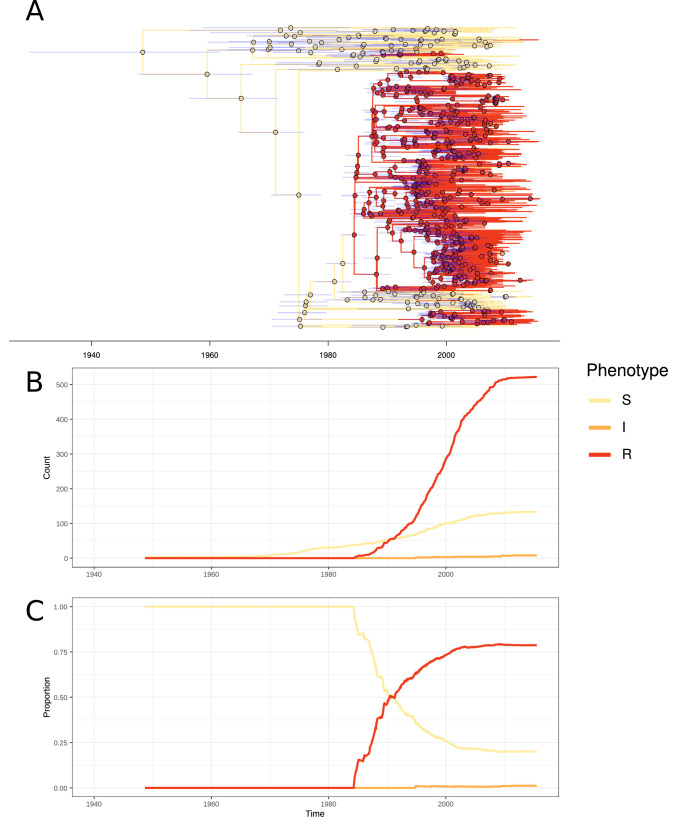
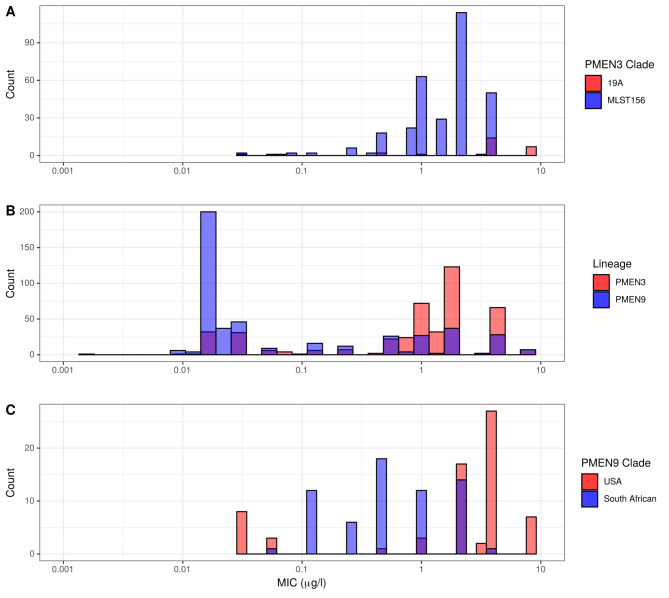
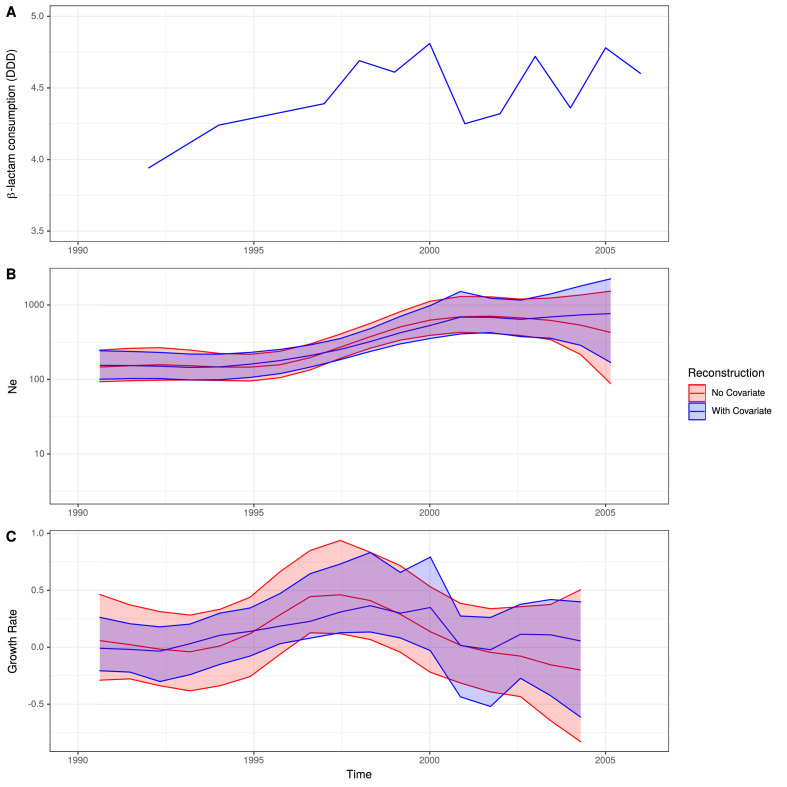
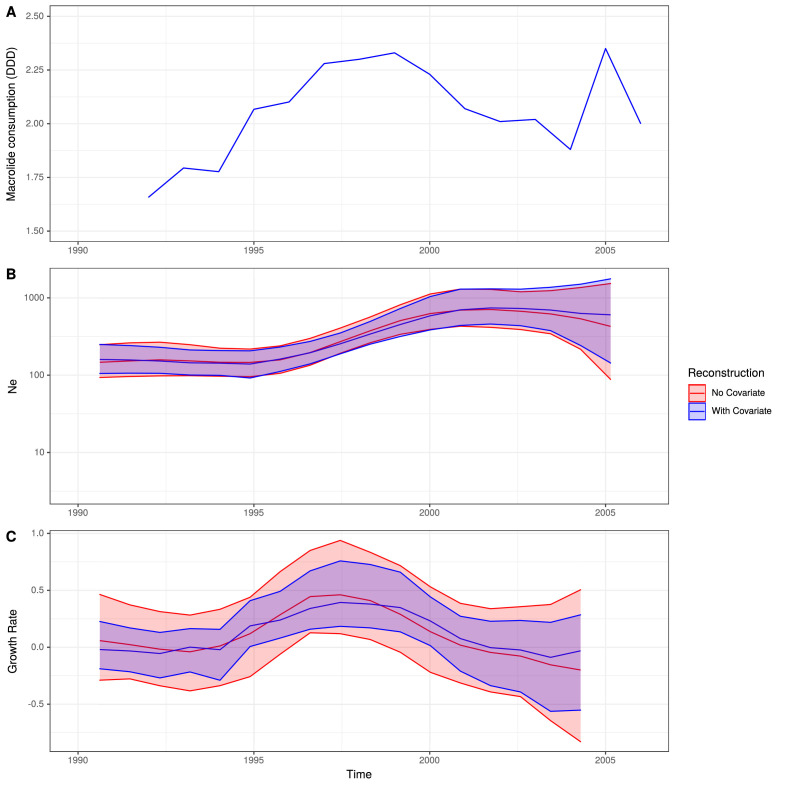
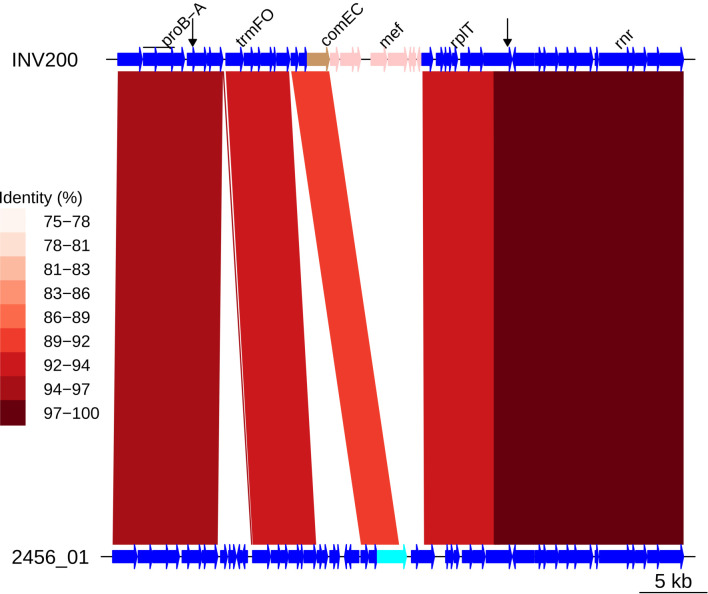
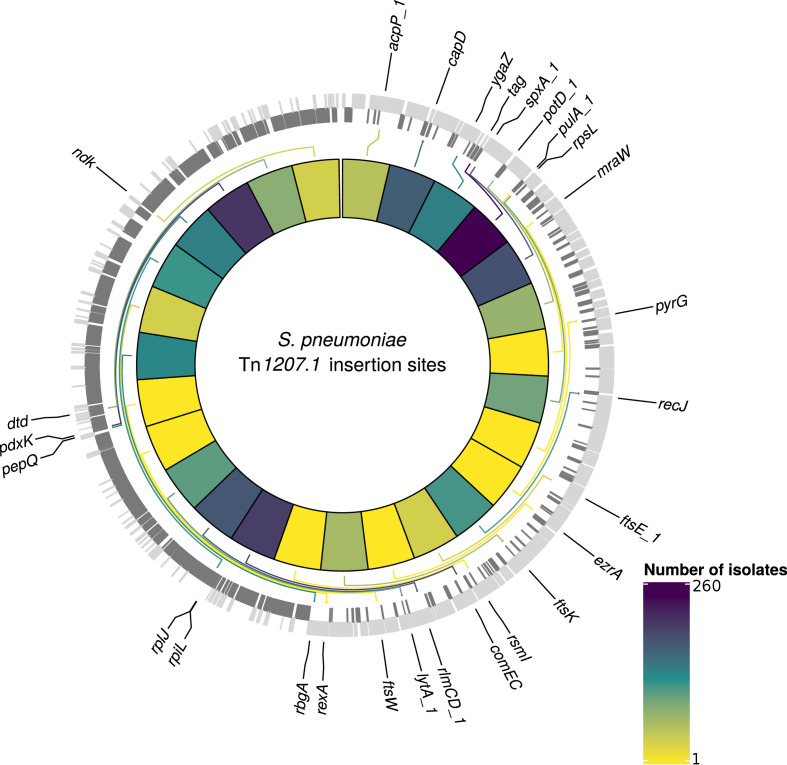
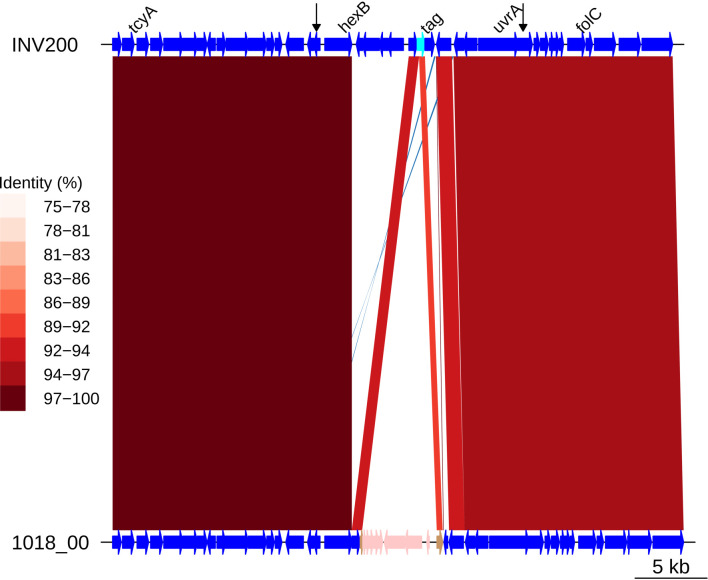
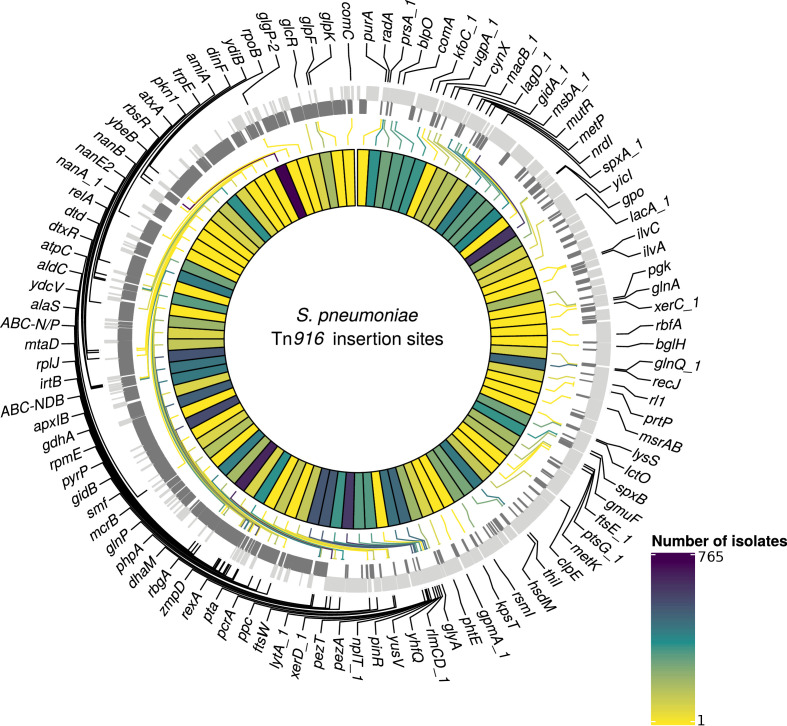
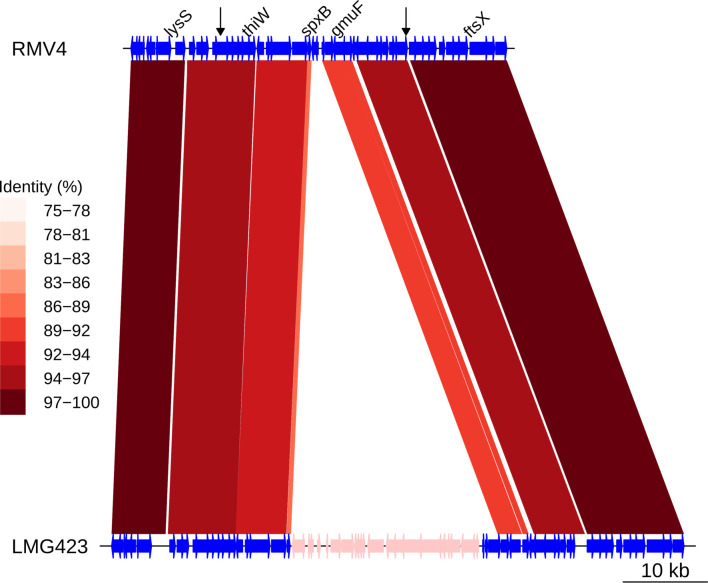
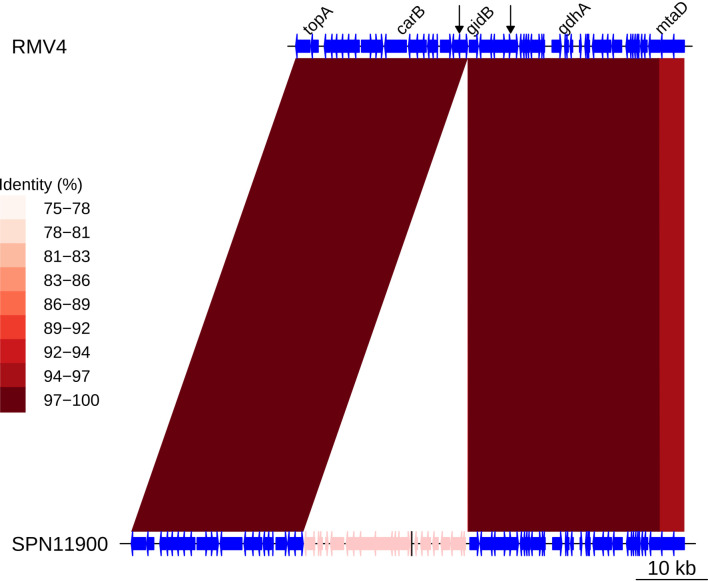
Multidrug-resistant Streptococcus pneumoniae emerge through the modification of core genome loci by interspecies homologous recombinations, and acquisition of gene cassettes. Both occurred in the otherwise contrasting histories of the antibiotic-resistant S. pneumoniae lineages PMEN3 and PMEN9. A single PMEN3 clade spread globally, evading vaccine-induced immunity through frequent serotype switching, whereas locally circulating PMEN9 clades independently gained resistance. Both lineages repeatedly integrated Tn916-type and Tn1207.1-type elements, conferring tetracycline and macrolide resistance, respectively, through homologous recombination importing sequences originating in other species. A species-wide dataset found over 100 instances of such interspecific acquisitions of resistance cassettes and flanking homologous arms. Phylodynamic analysis of the most commonly sampled Tn1207.1-type insertion in PMEN9, originating from a commensal and disrupting a competence gene, suggested its expansion across Germany was driven by a high ratio of macrolide-to-β-lactam consumption. Hence, selection from antibiotic consumption was sufficient for these atypically large recombinations to overcome species boundaries across the pneumococcal chromosome.
Keywords: AMR; epidemiology; genetics; genomics; infectious disease; microbiology; recombination; streptococcus pneumoniae.
Conflict of interest statement
JD, Mv, LM, Hd, PT, JS, SL, RG, RS, KK, WH, RB, BB, SB No competing interests declared, NC has consulted for Antigen Discovery Inc Has received an investigator-initiated award from GlaxoSmithKline.
Figures


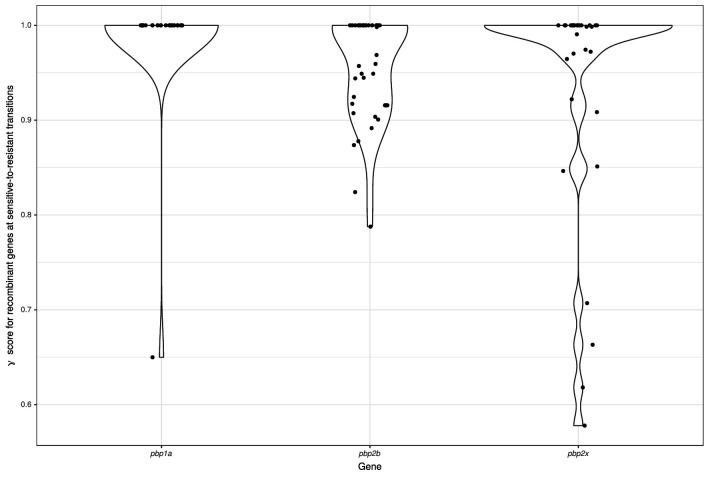
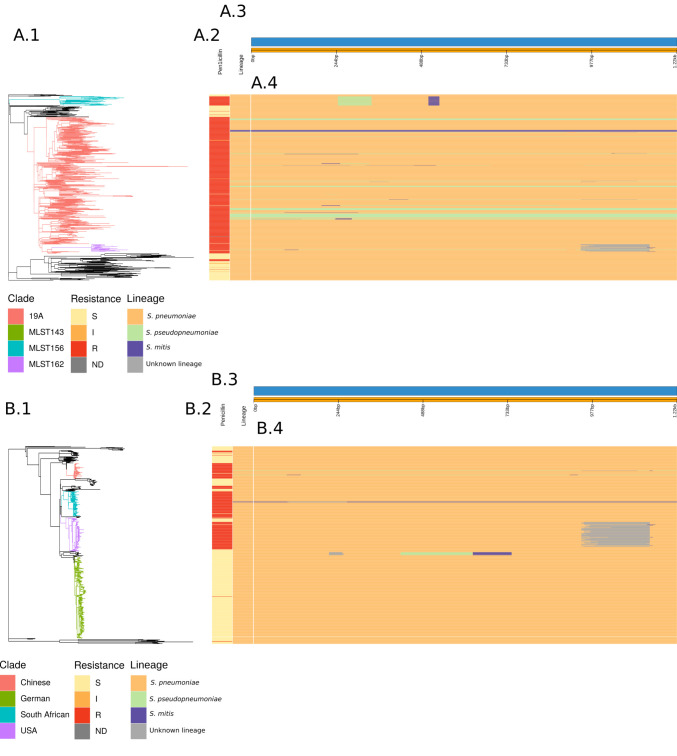
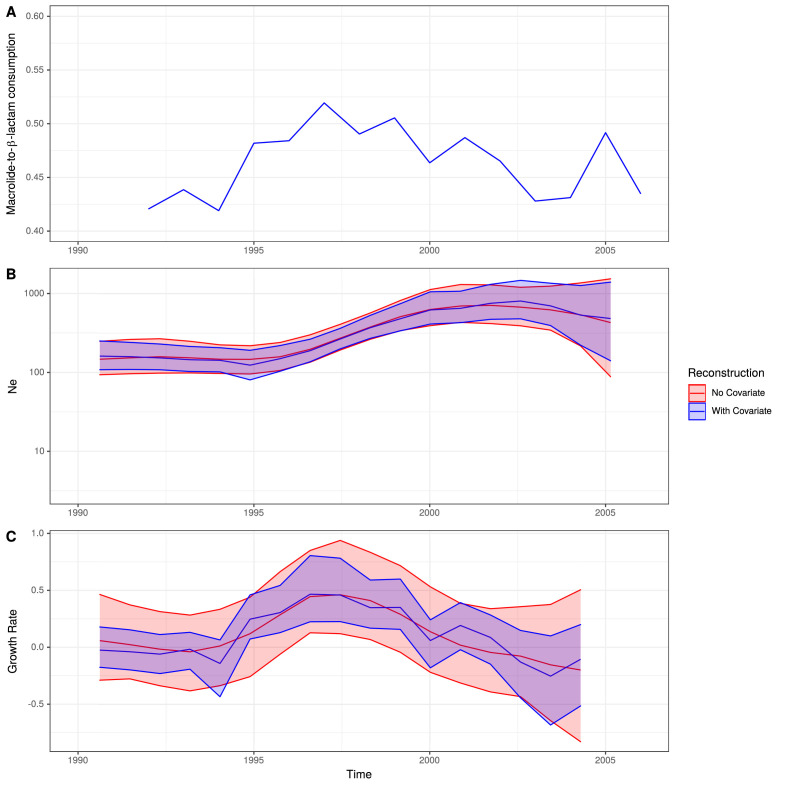
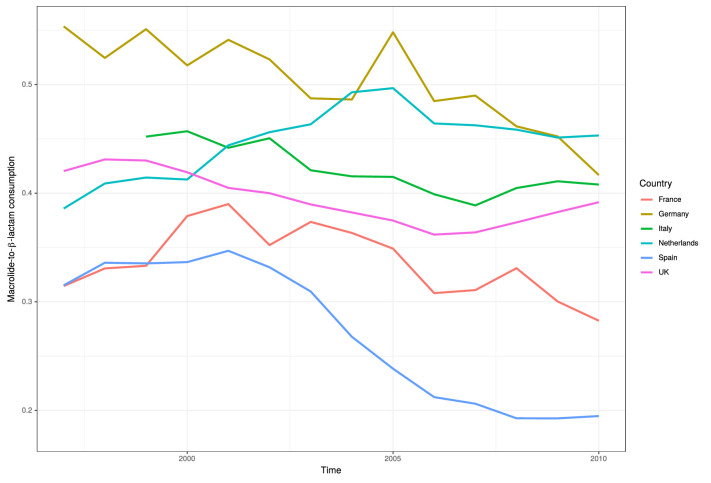


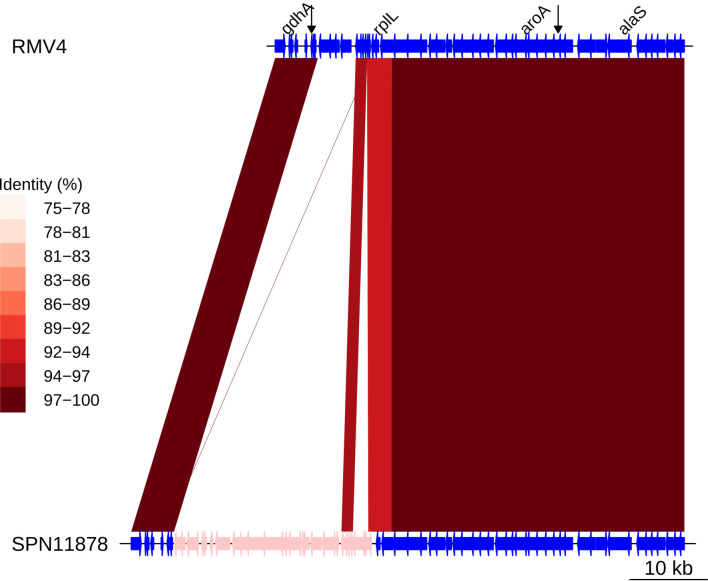
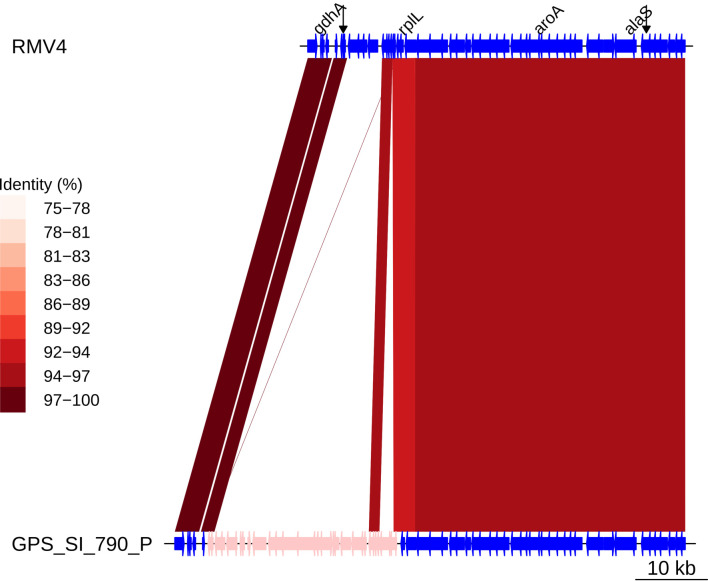

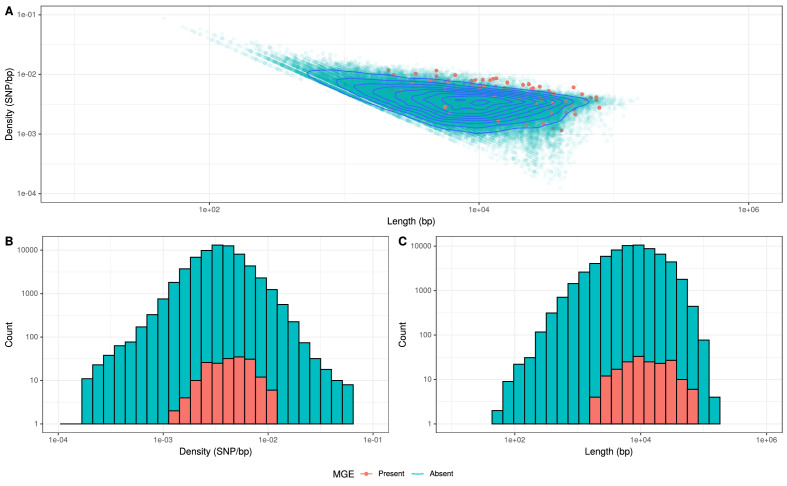
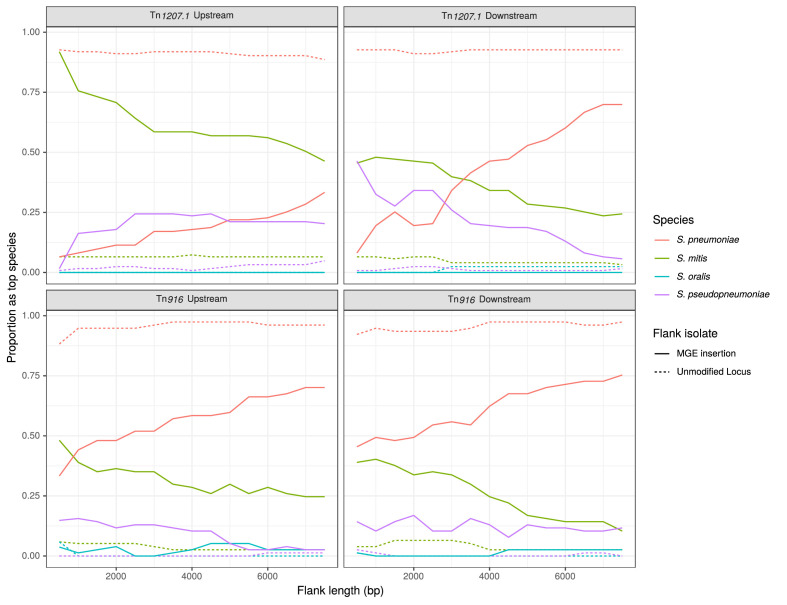
References
-
- Baker KS, Dallman TJ, Field N, Childs T, Mitchell H, Day M, Weill F-X, Lefèvre S, Tourdjman M, Hughes G, Jenkins C, Thomson N. Horizontal antimicrobial resistance transfer drives epidemics of multiple Shigella species. Nature Communications. 2018;9:1462. doi: 10.1038/s41467-018-03949-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
