

COVID-19 virtual patient cohort suggests immune mechanisms driving disease outcomes
- PMID: 34260666
- PMCID: PMC8312984
- DOI: 10.1371/journal.ppat.1009753
COVID-19 virtual patient cohort suggests immune mechanisms driving disease outcomes
Abstract
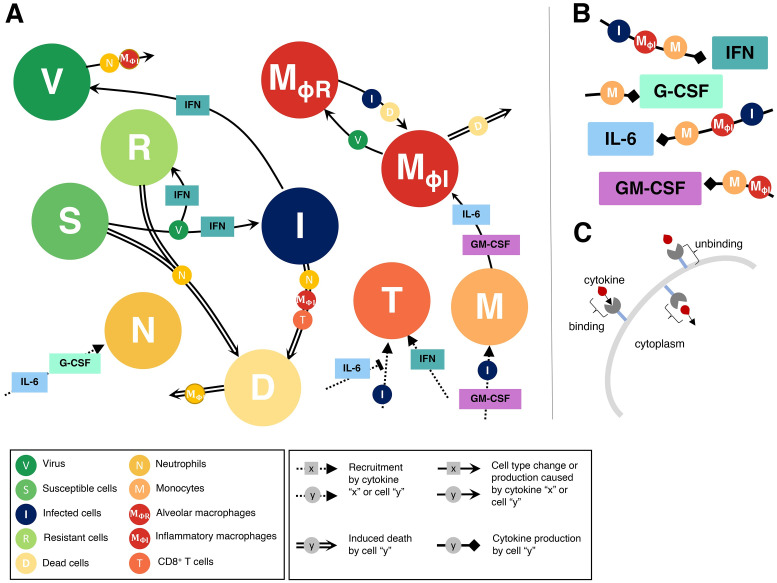
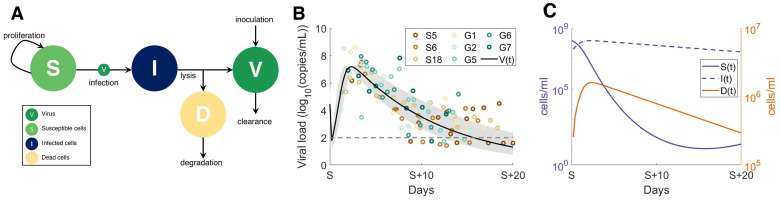
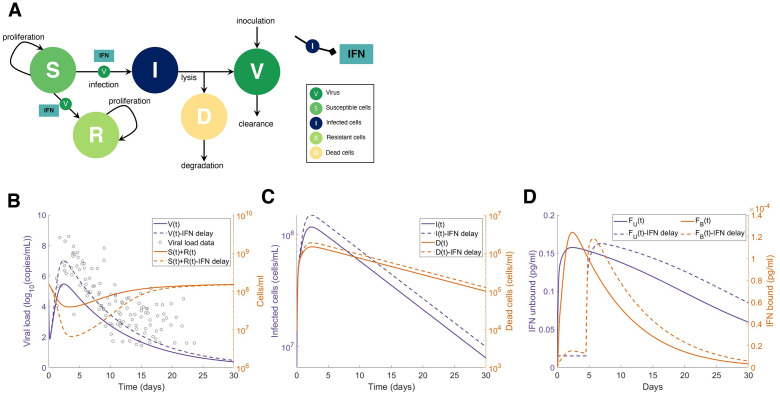
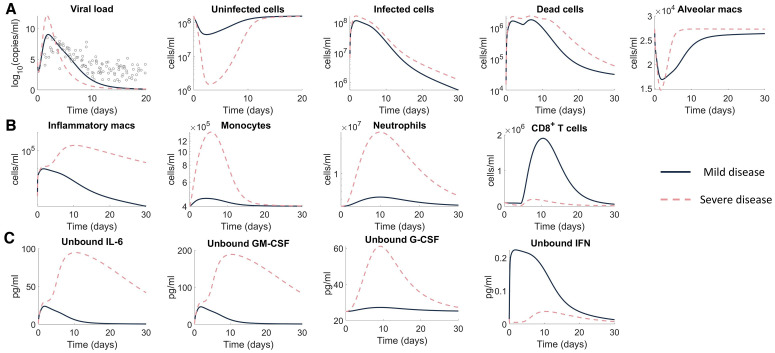
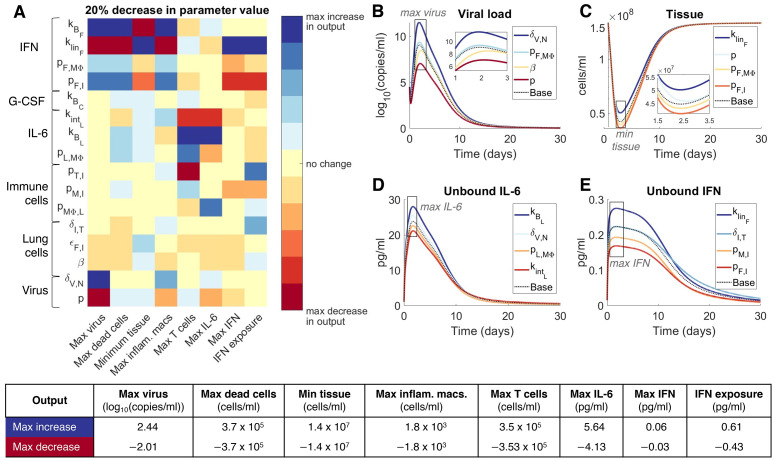
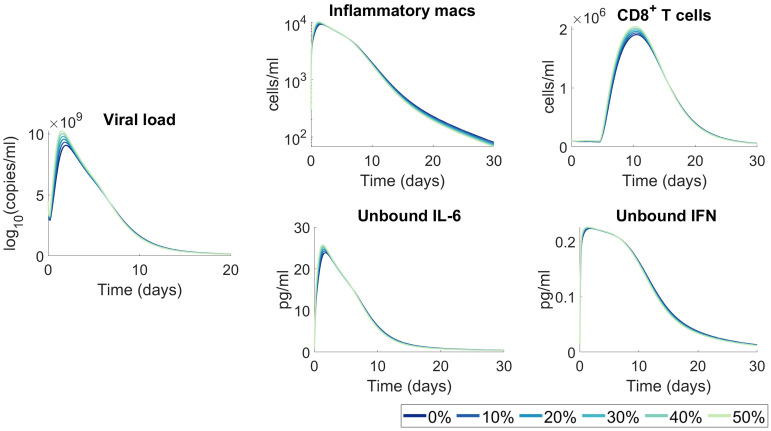
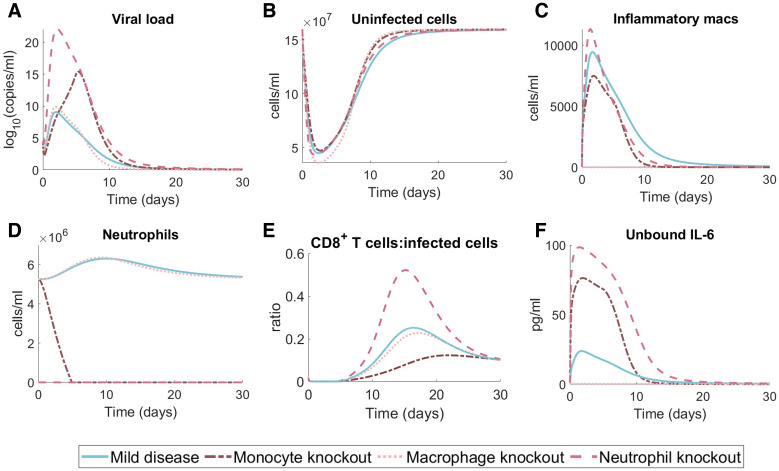
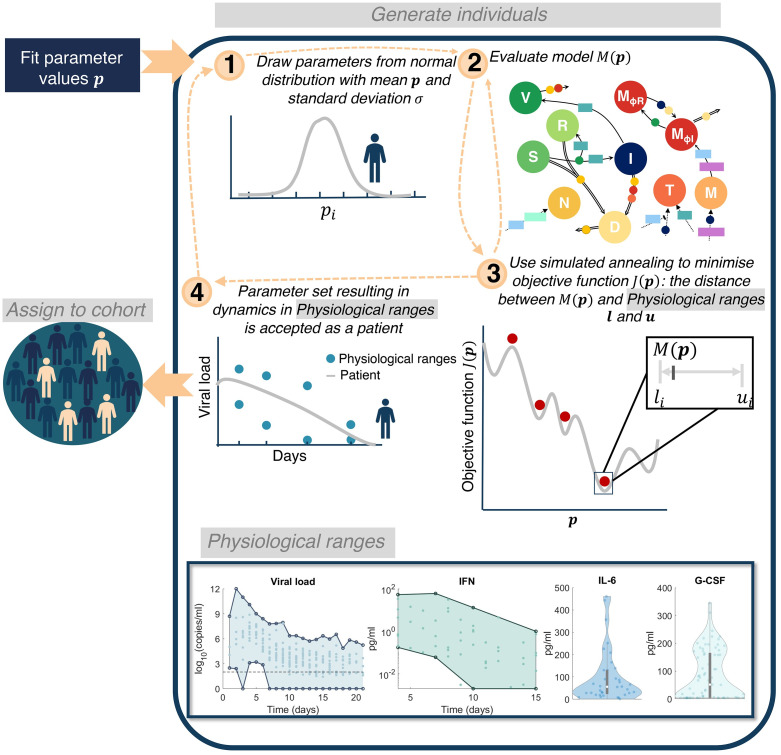
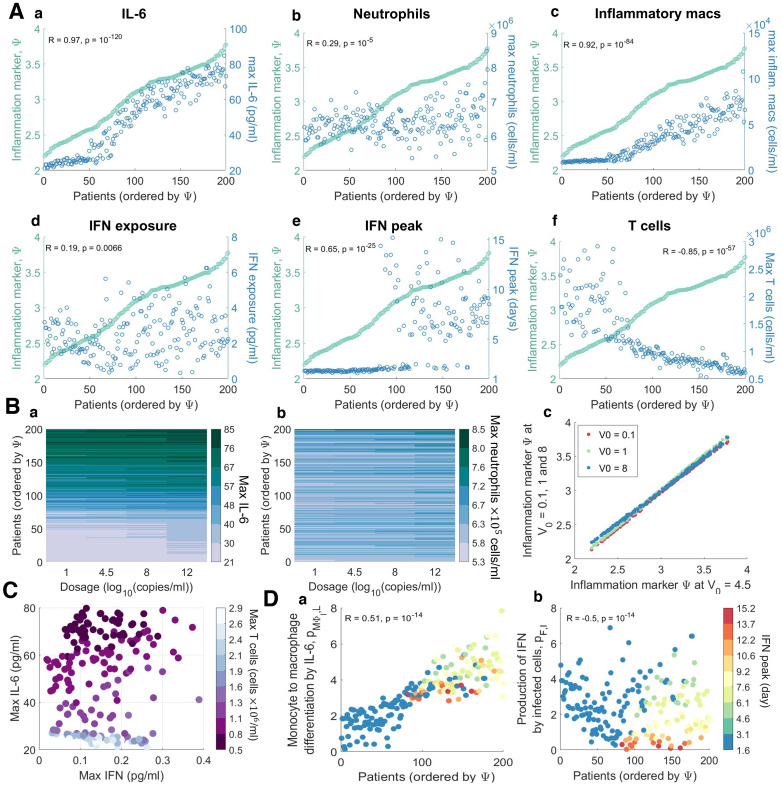
To understand the diversity of immune responses to SARS-CoV-2 and distinguish features that predispose individuals to severe COVID-19, we developed a mechanistic, within-host mathematical model and virtual patient cohort. Our results suggest that virtual patients with low production rates of infected cell derived IFN subsequently experienced highly inflammatory disease phenotypes, compared to those with early and robust IFN responses. In these in silico patients, the maximum concentration of IL-6 was also a major predictor of CD8+ T cell depletion. Our analyses predicted that individuals with severe COVID-19 also have accelerated monocyte-to-macrophage differentiation mediated by increased IL-6 and reduced type I IFN signalling. Together, these findings suggest biomarkers driving the development of severe COVID-19 and support early interventions aimed at reducing inflammation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Update of
-
COVID-19 virtual patient cohort reveals immune mechanisms driving disease outcomes.bioRxiv [Preprint]. 2021 Jan 6:2021.01.05.425420. doi: 10.1101/2021.01.05.425420. bioRxiv. 2021. Update in: PLoS Pathog. 2021 Jul 14;17(7):e1009753. doi: 10.1371/journal.ppat.1009753. PMID: 33442689 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
