

A metabolomics pipeline for the mechanistic interrogation of the gut microbiome
- PMID: 34262212
- PMCID: PMC8939302
- DOI: 10.1038/s41586-021-03707-9
A metabolomics pipeline for the mechanistic interrogation of the gut microbiome
Abstract
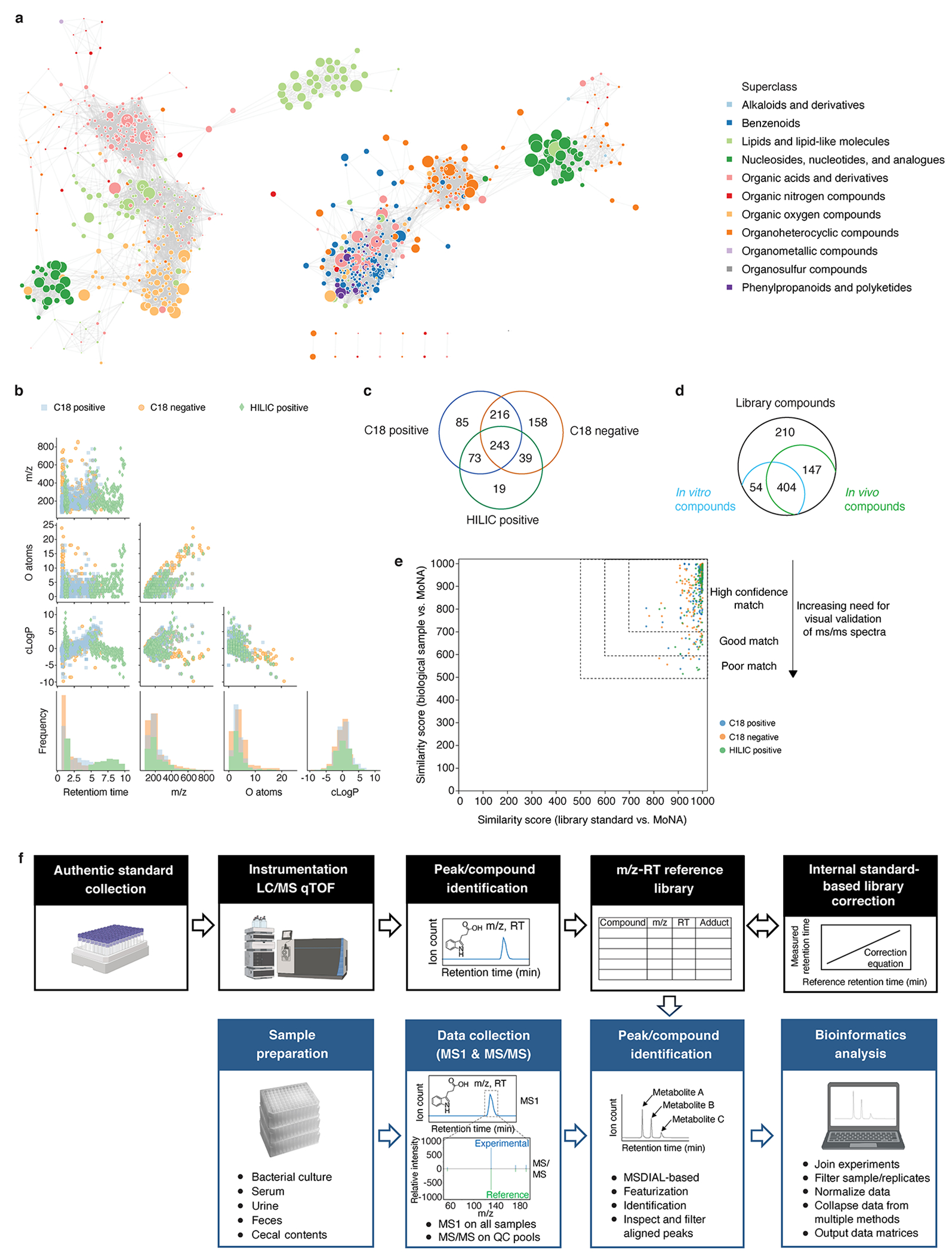
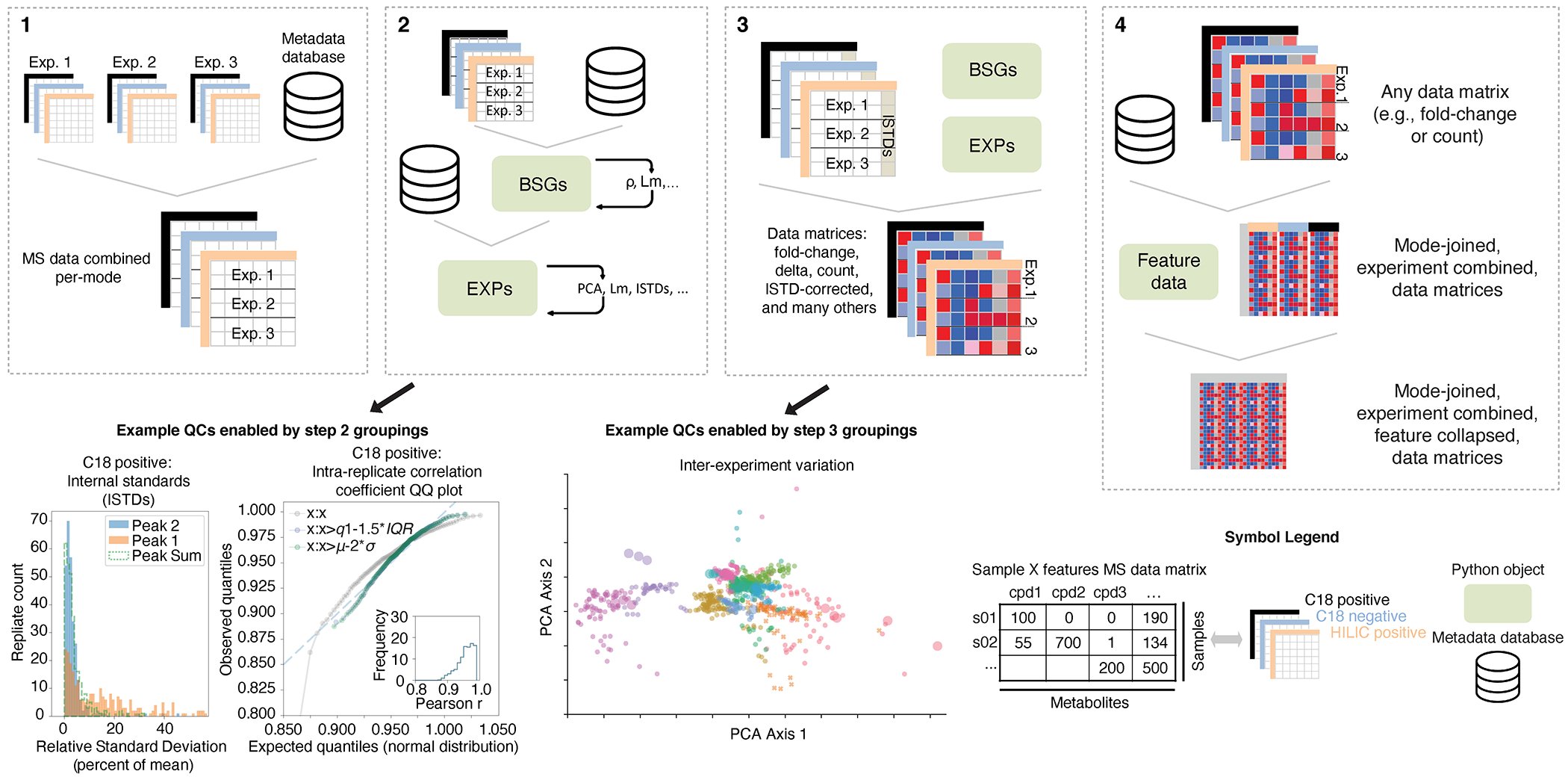
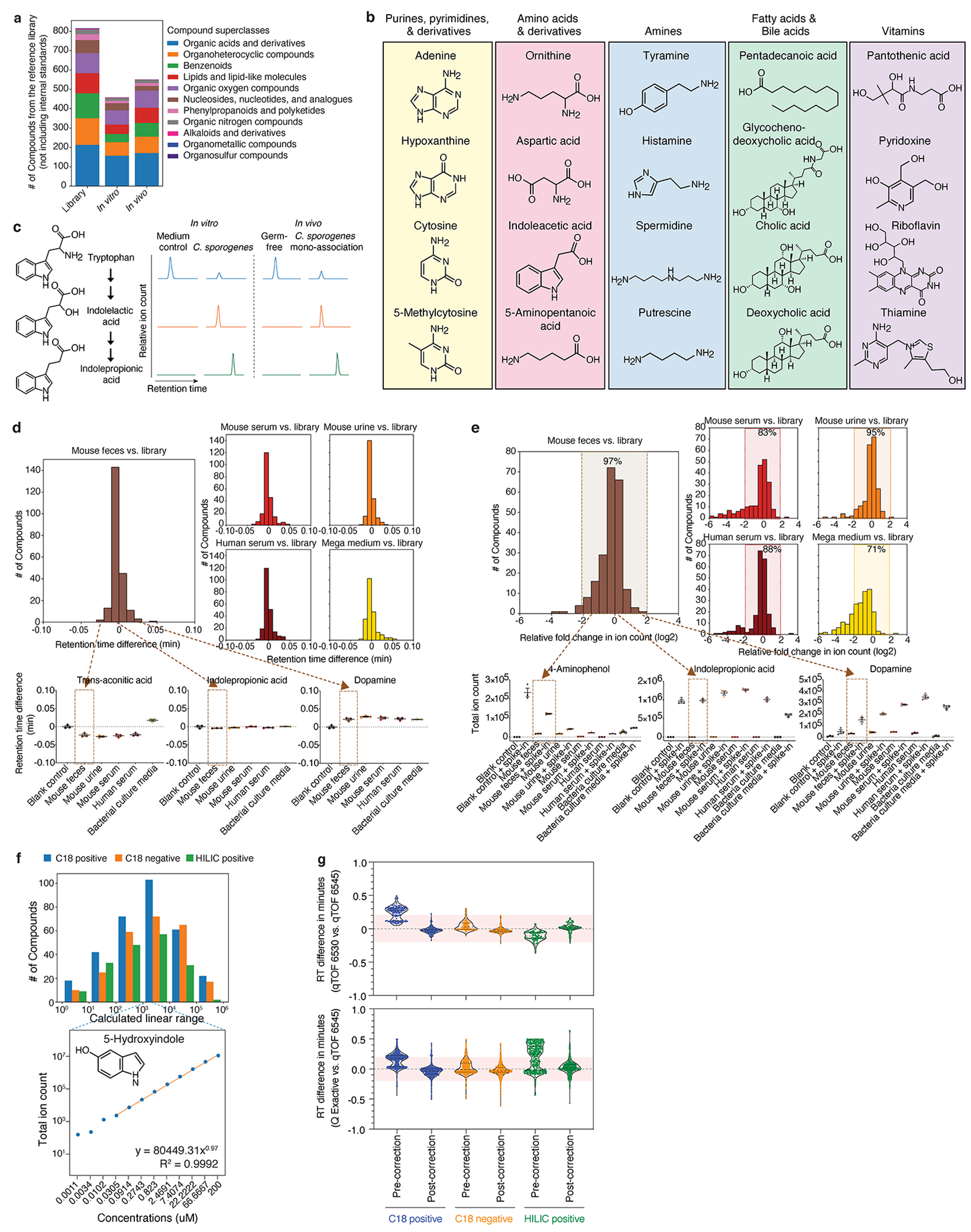
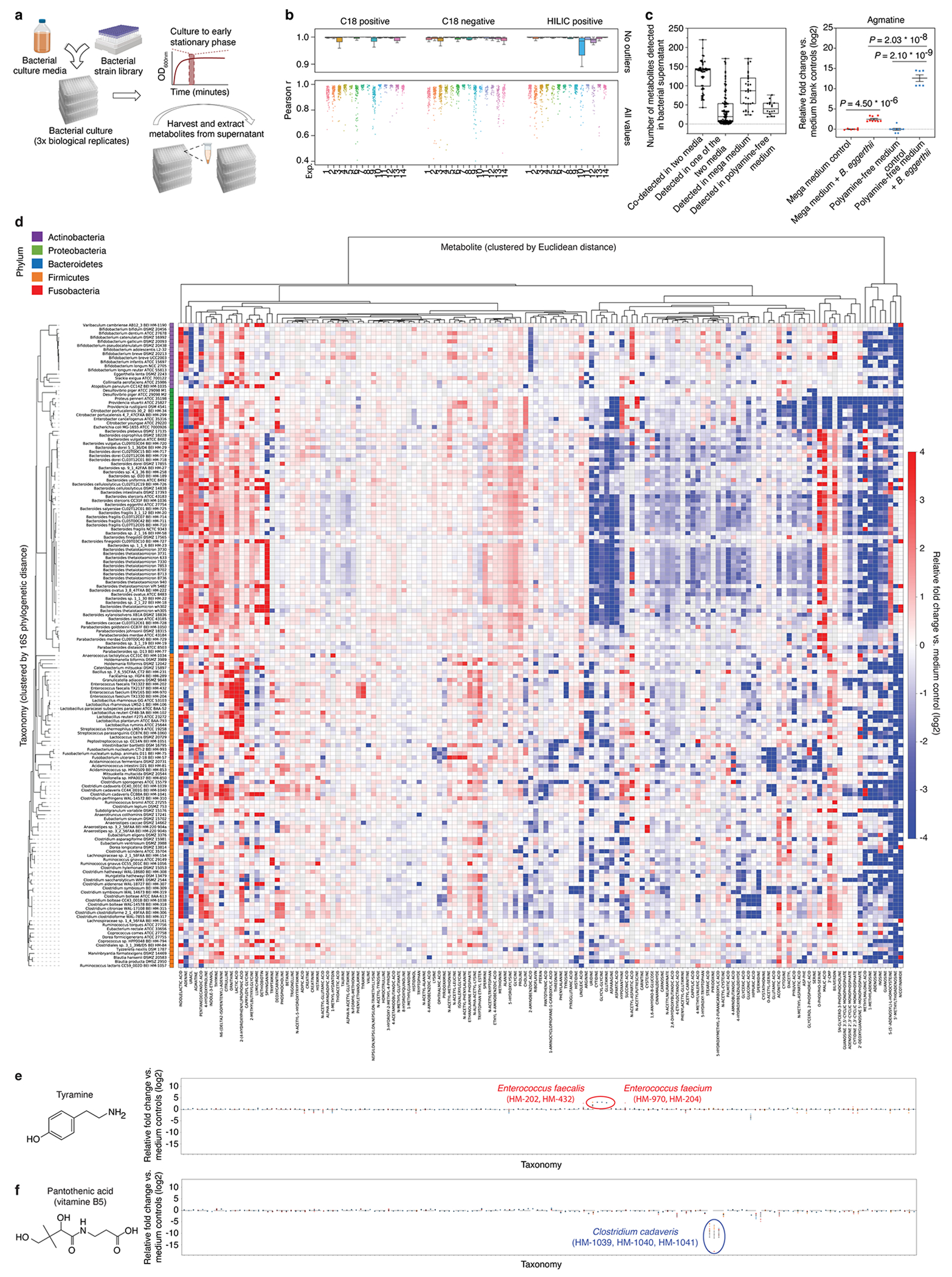
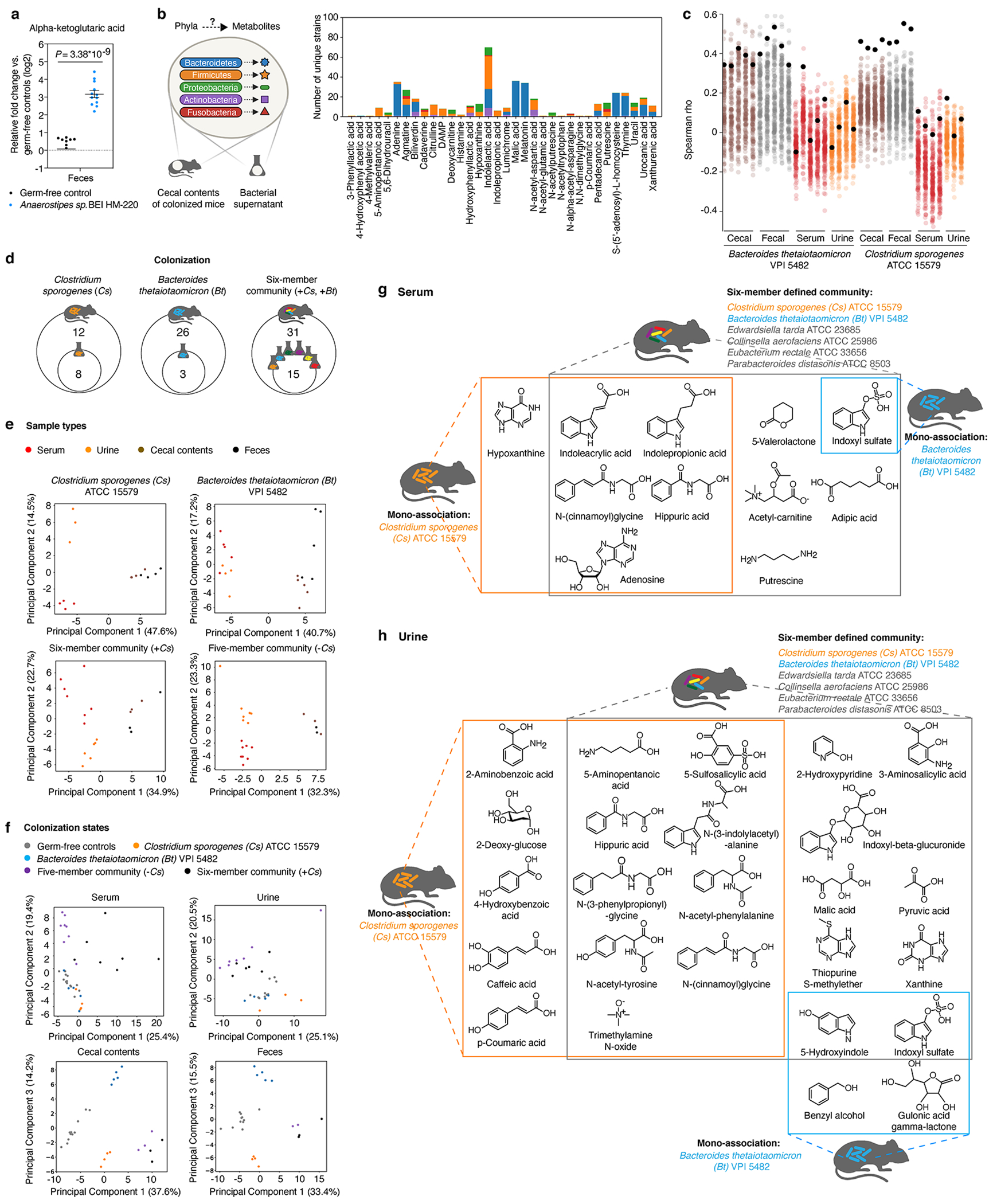
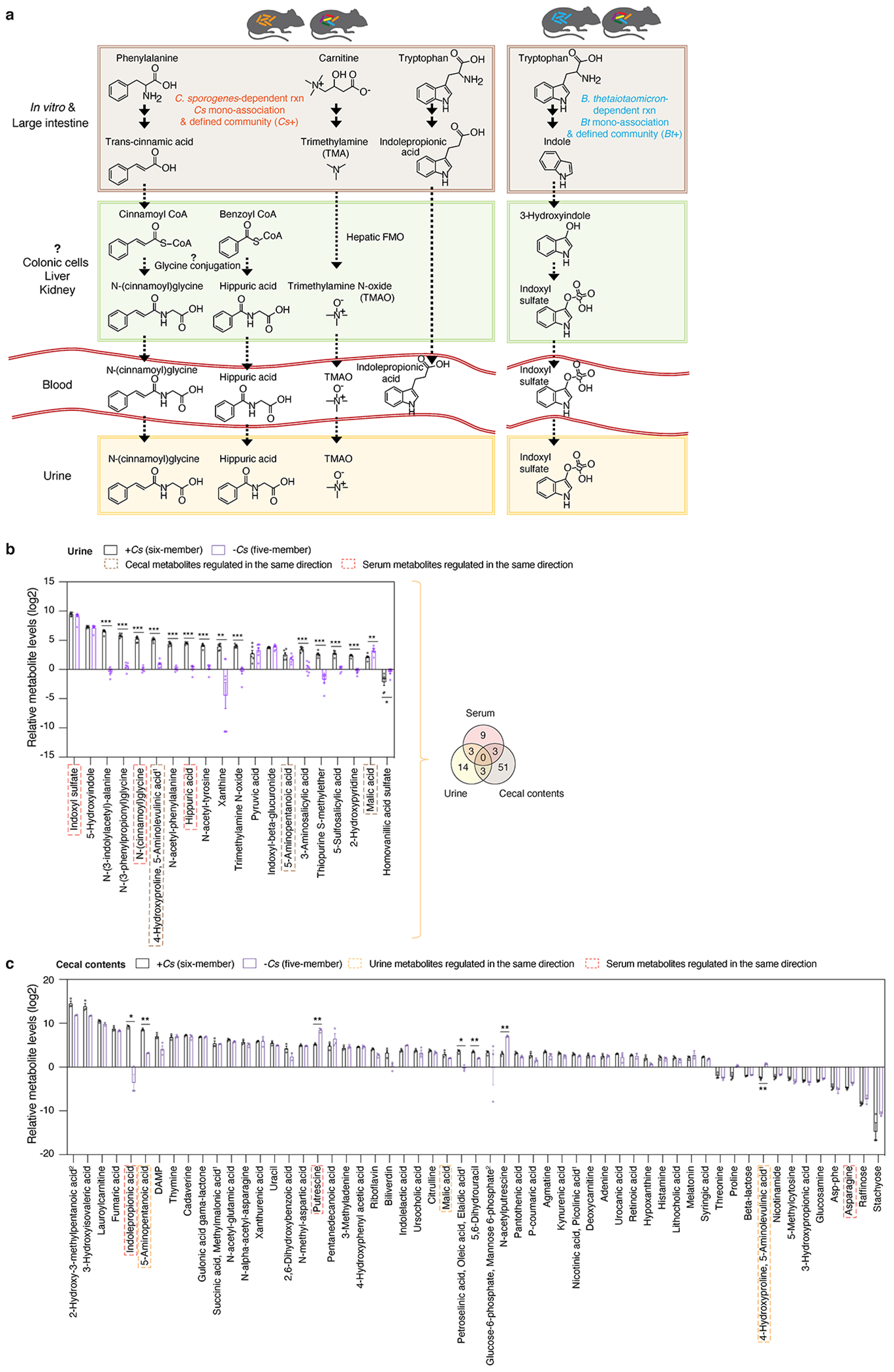
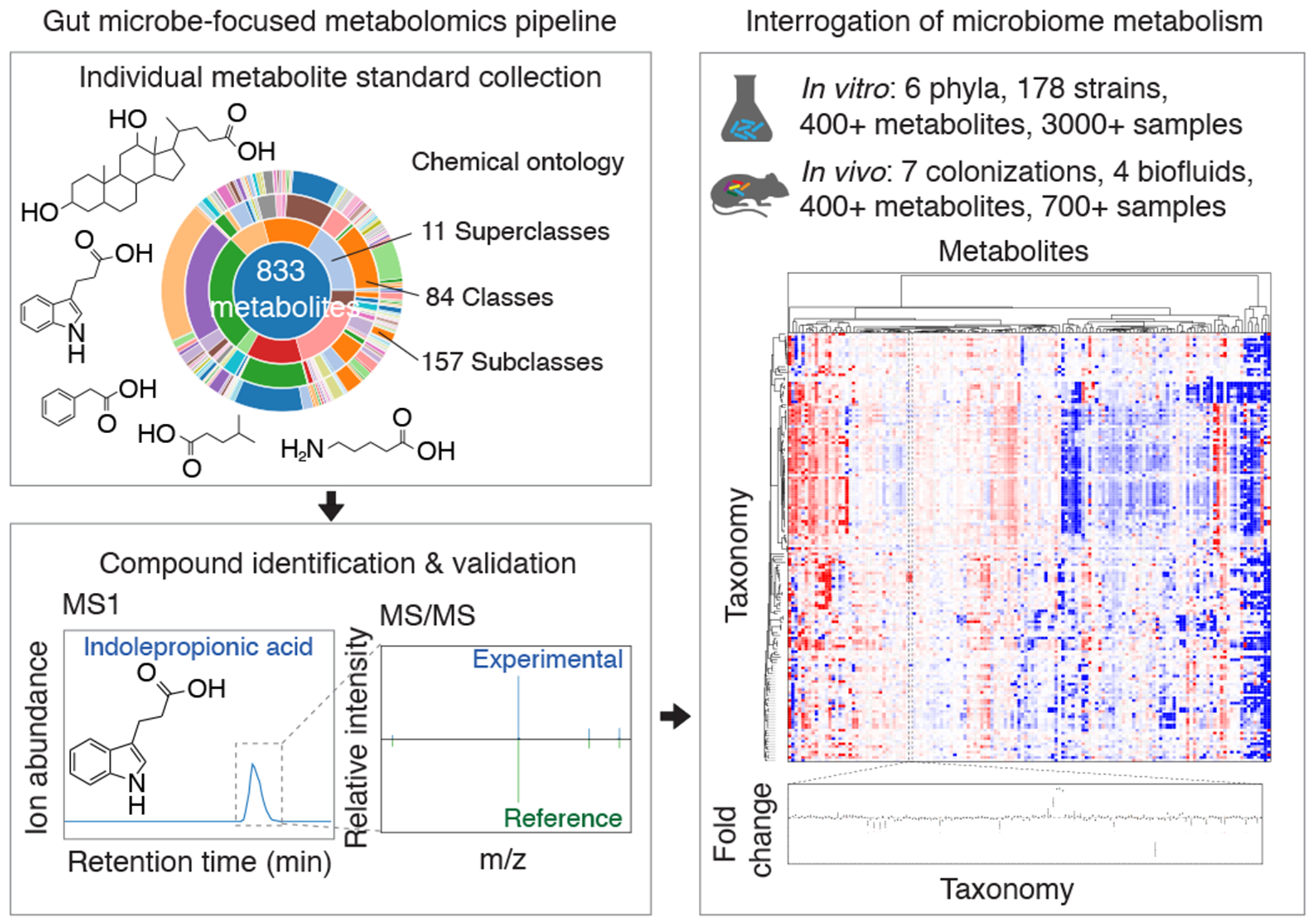
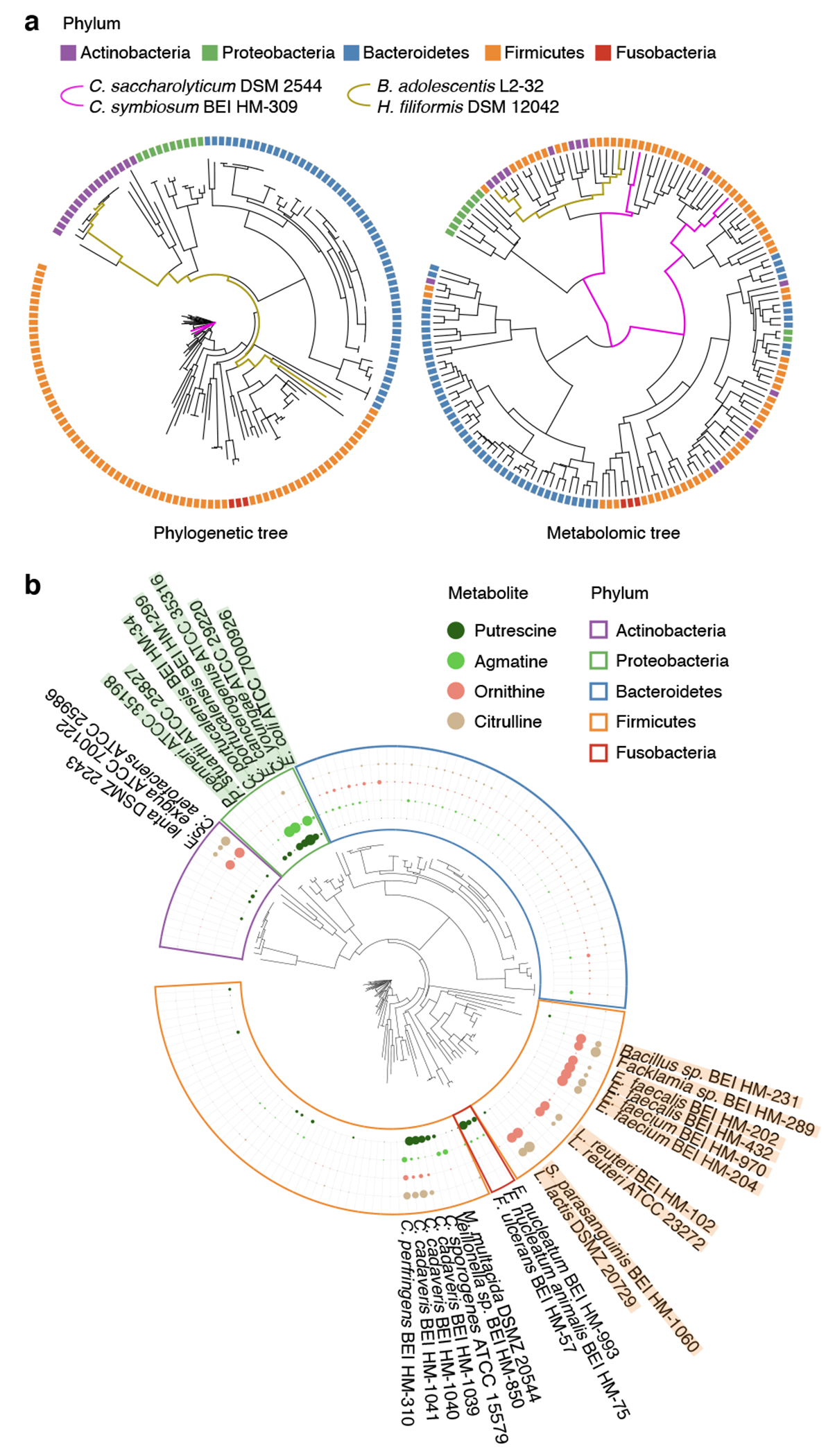
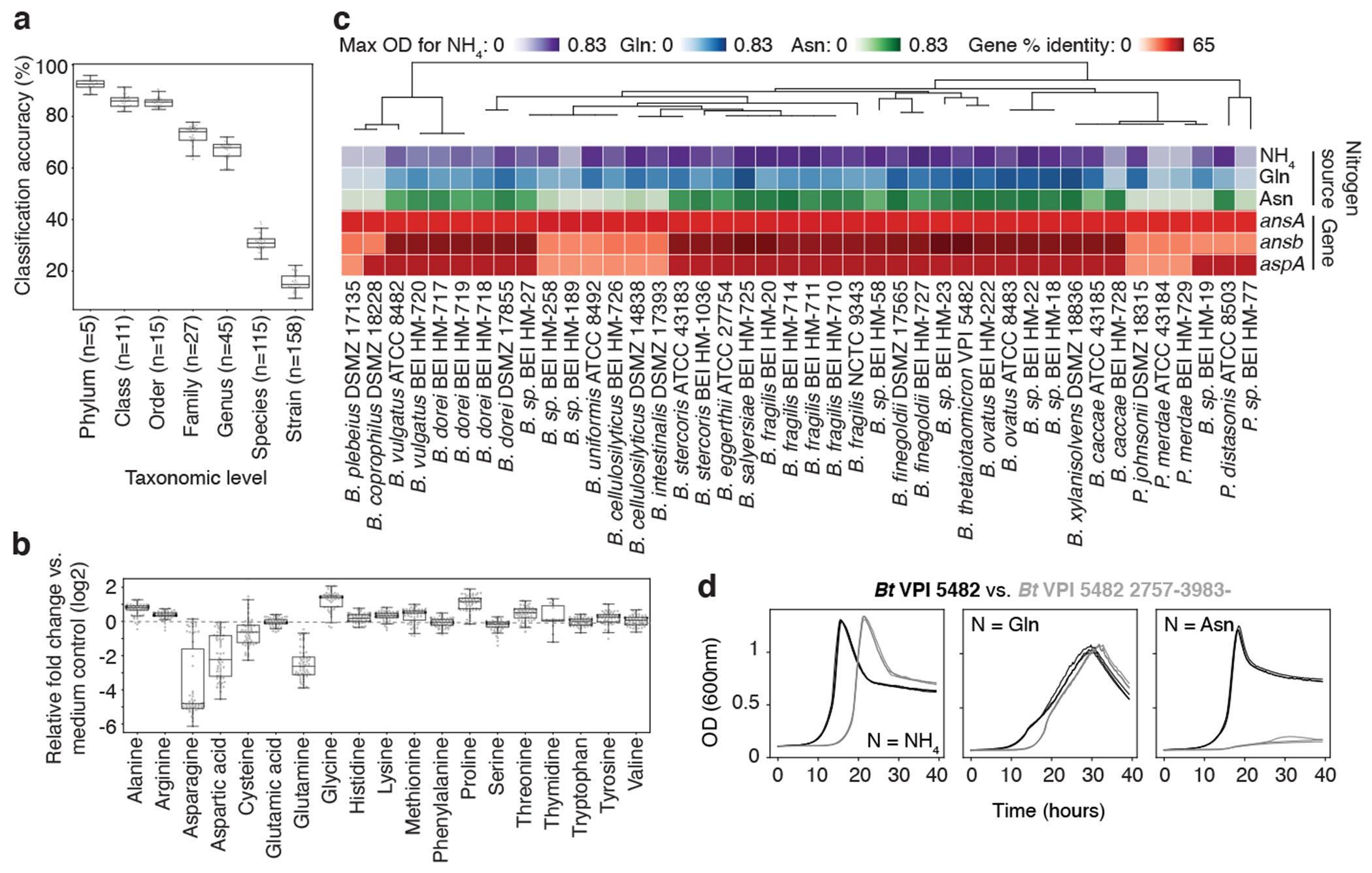
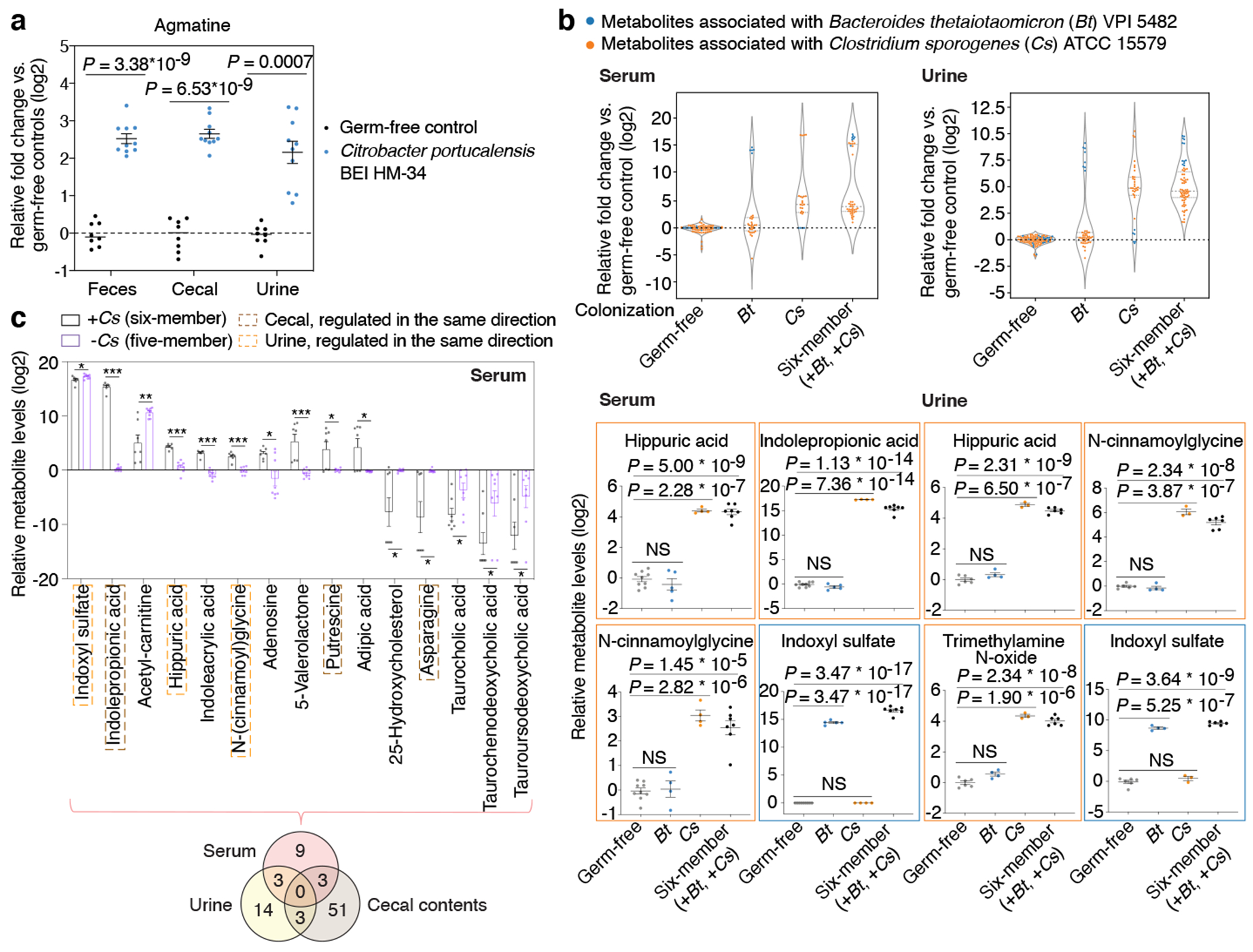
Gut microorganisms modulate host phenotypes and are associated with numerous health effects in humans, ranging from host responses to cancer immunotherapy to metabolic disease and obesity. However, difficulty in accurate and high-throughput functional analysis of human gut microorganisms has hindered efforts to define mechanistic connections between individual microbial strains and host phenotypes. One key way in which the gut microbiome influences host physiology is through the production of small molecules1-3, yet progress in elucidating this chemical interplay has been hindered by limited tools calibrated to detect the products of anaerobic biochemistry in the gut. Here we construct a microbiome-focused, integrated mass-spectrometry pipeline to accelerate the identification of microbiota-dependent metabolites in diverse sample types. We report the metabolic profiles of 178 gut microorganism strains using our library of 833 metabolites. Using this metabolomics resource, we establish deviations in the relationships between phylogeny and metabolism, use machine learning to discover a previously undescribed type of metabolism in Bacteroides, and reveal candidate biochemical pathways using comparative genomics. Microbiota-dependent metabolites can be detected in diverse biological fluids from gnotobiotic and conventionally colonized mice and traced back to the corresponding metabolomic profiles of cultured bacteria. Collectively, our microbiome-focused metabolomics pipeline and interactive metabolomics profile explorer are a powerful tool for characterizing microorganisms and interactions between microorganisms and their host.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures
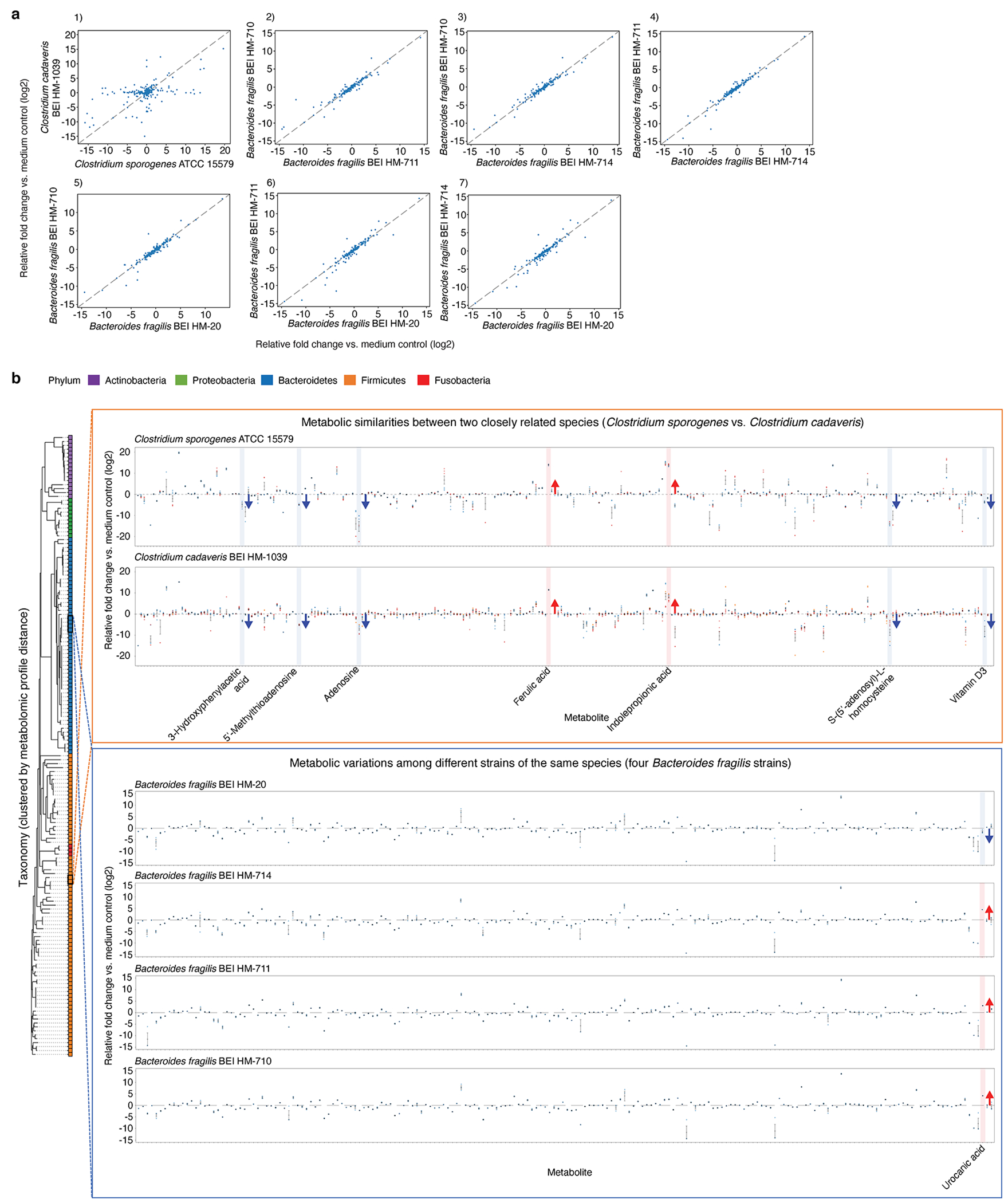
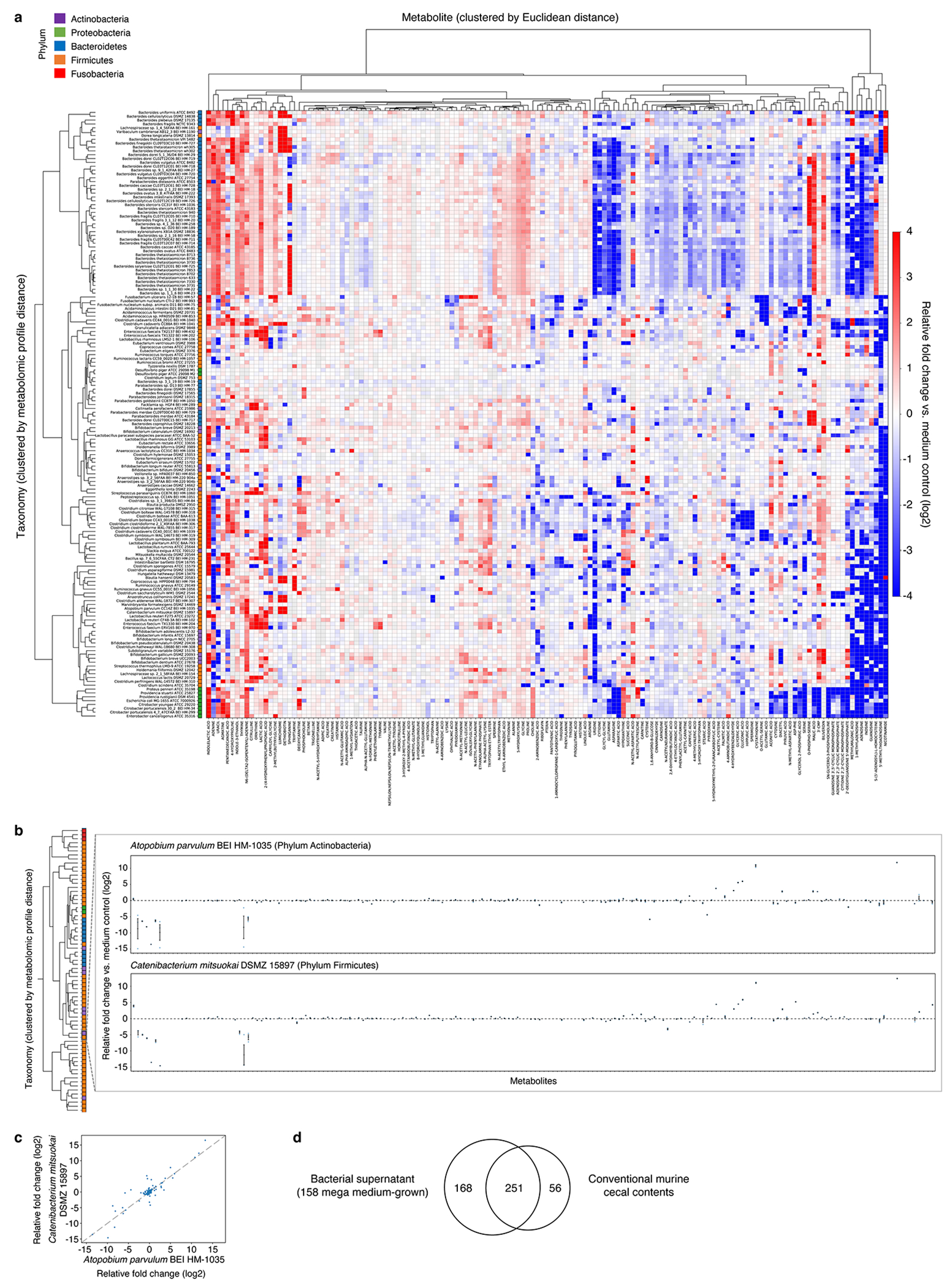
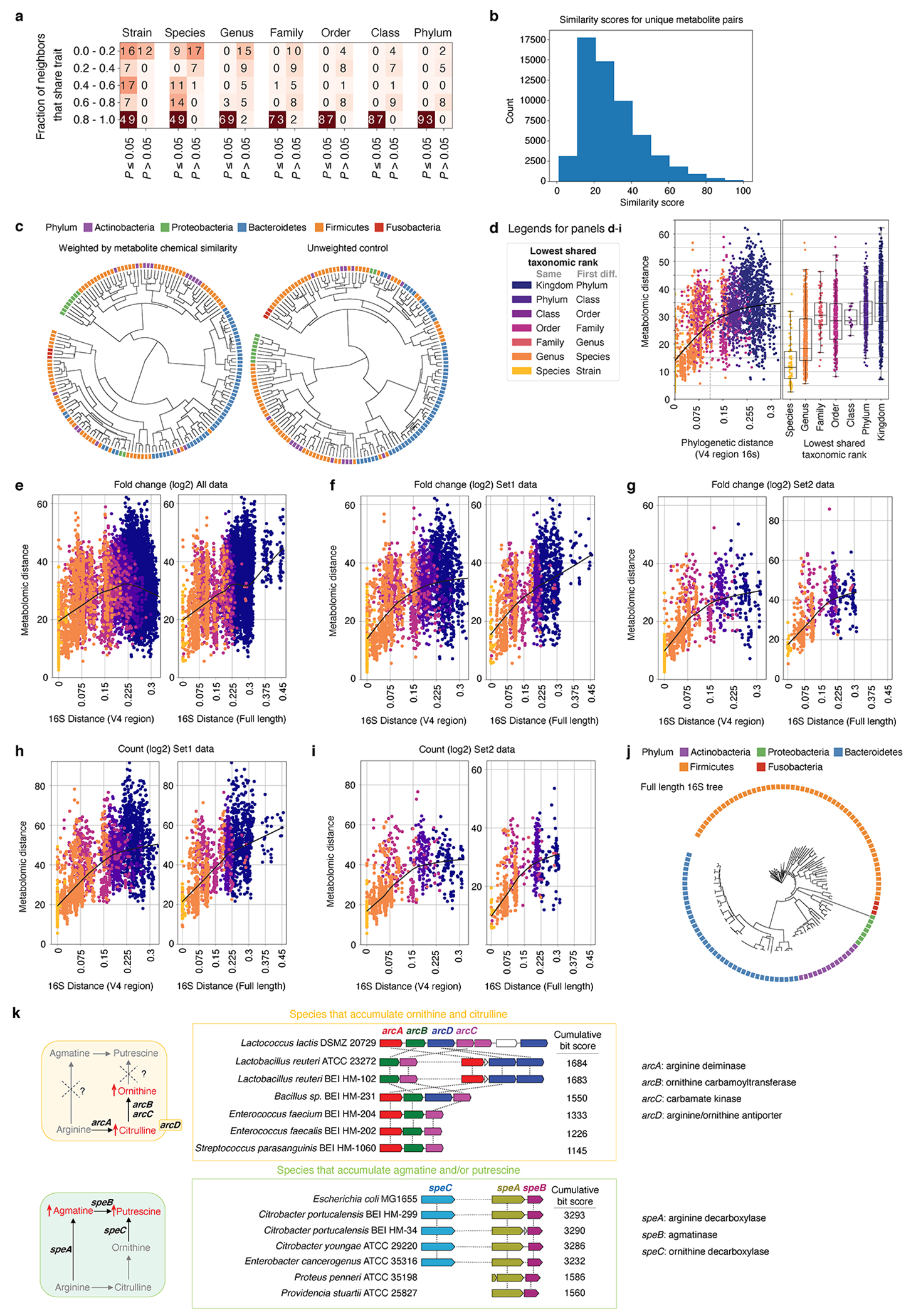
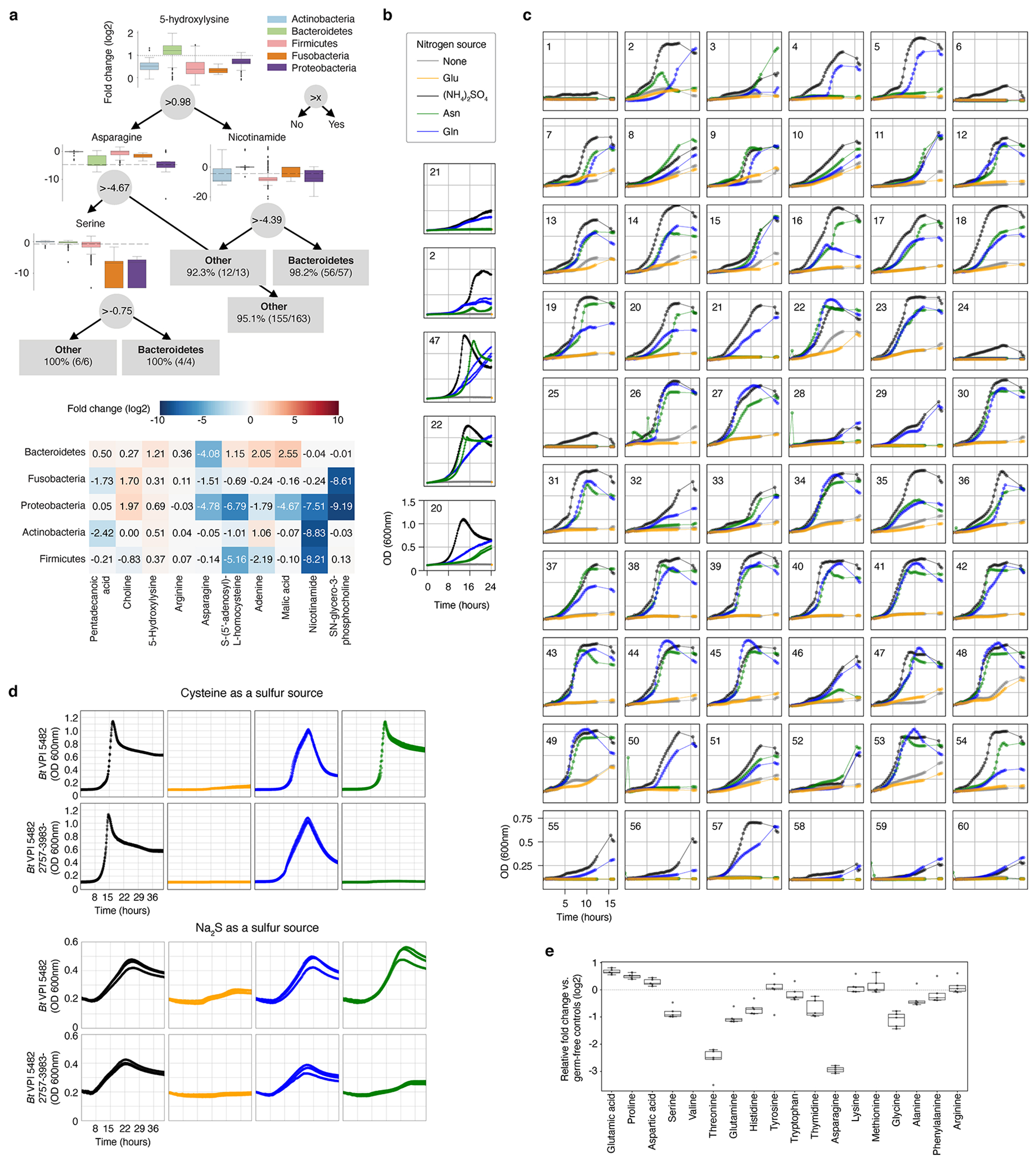
Comment in
-
Deciphering metabolism, one microbe at a time.Nature. 2021 Jul;595(7867):355-357. doi: 10.1038/d41586-021-01774-6. Nature. 2021. PMID: 34262197 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
