

Assessing conservation of alternative splicing with evolutionary splicing graphs
- PMID: 34266979
- PMCID: PMC8327911
- DOI: 10.1101/gr.274696.120
Assessing conservation of alternative splicing with evolutionary splicing graphs
Abstract
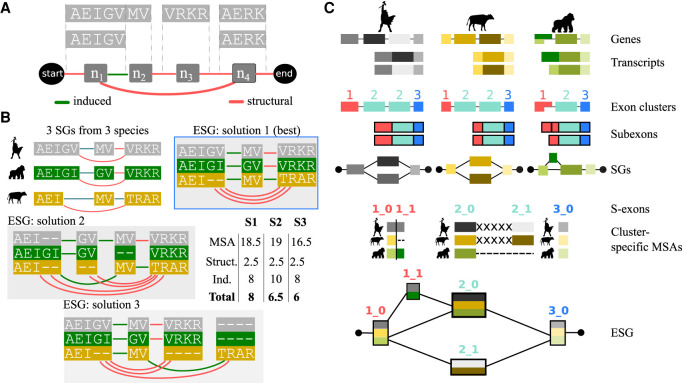
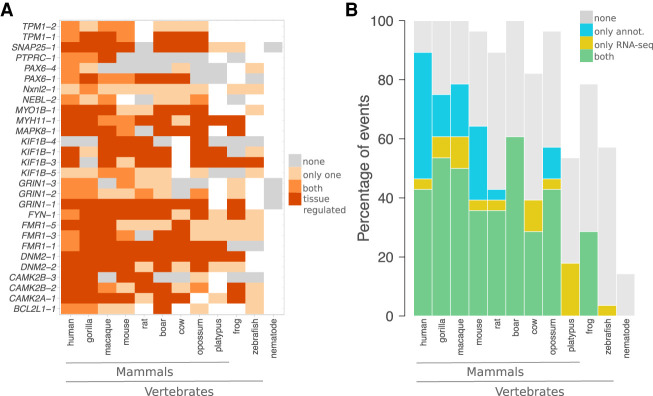
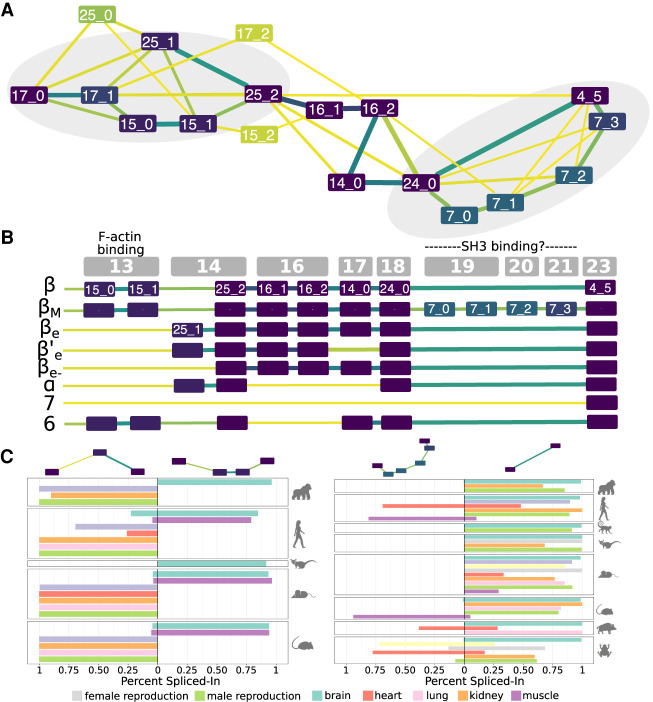
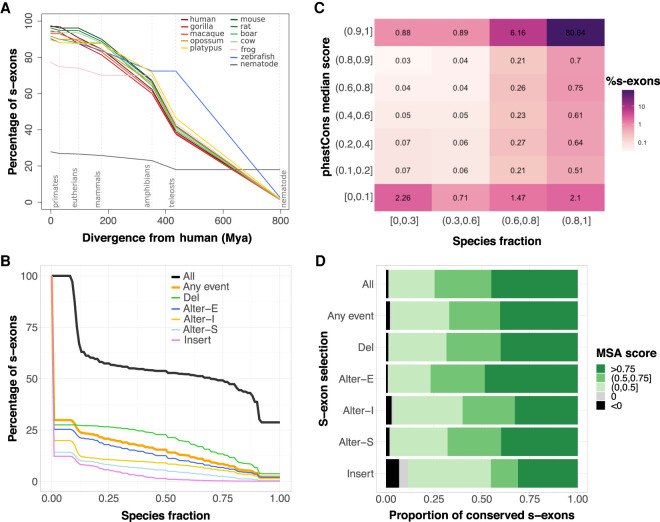
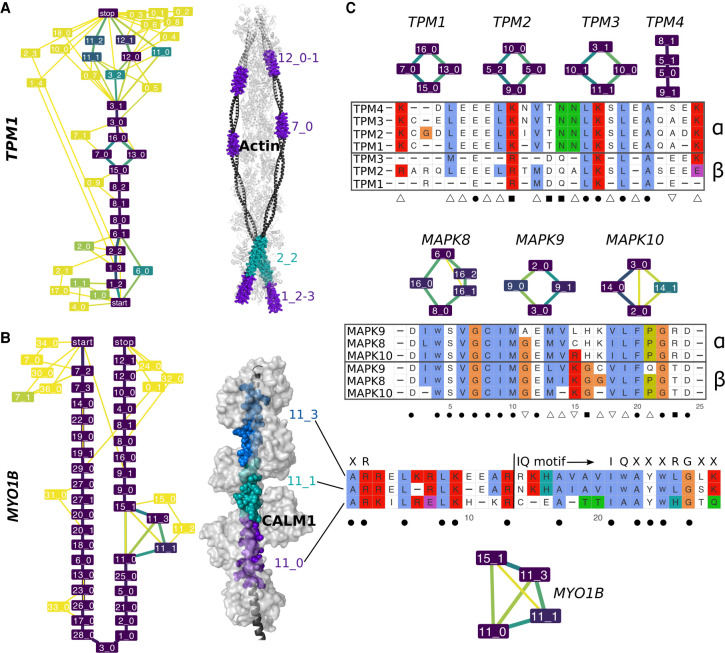
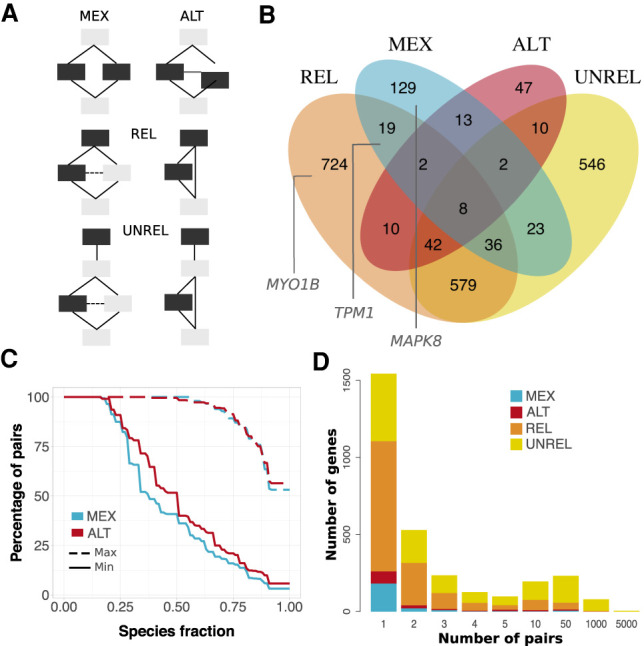
Understanding how protein function has evolved and diversified is of great importance for human genetics and medicine. Here, we tackle the problem of describing the whole transcript variability observed in several species by generalizing the definition of splicing graph. We provide a practical solution to construct parsimonious evolutionary splicing graphs where each node is a minimal transcript building block defined across species. We show a clear link between the functional relevance, tissue regulation, and conservation of alternative transcripts on a set of 50 genes. By scaling up to the whole human protein-coding genome, we identify a few thousand genes where alternative splicing modulates the number and composition of pseudorepeats. We have implemented our approach in ThorAxe, an efficient, versatile, robust, and freely available computational tool.
© 2021 Zea et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources