

3Cnet: pathogenicity prediction of human variants using multitask learning with evolutionary constraints
- PMID: 34270679
- PMCID: PMC8665754
- DOI: 10.1093/bioinformatics/btab529
3Cnet: pathogenicity prediction of human variants using multitask learning with evolutionary constraints
Abstract
Motivation: Improvements in next-generation sequencing have enabled genome-based diagnosis for patients with genetic diseases. However, accurate interpretation of human variants requires knowledge from a number of clinical cases. In addition, manual analysis of each variant detected in a patient's genome requires enormous time and effort. To reduce the cost of diagnosis, various computational tools have been developed to predict the pathogenicity of human variants, but the shortage and bias of available clinical data can lead to overfitting of algorithms.
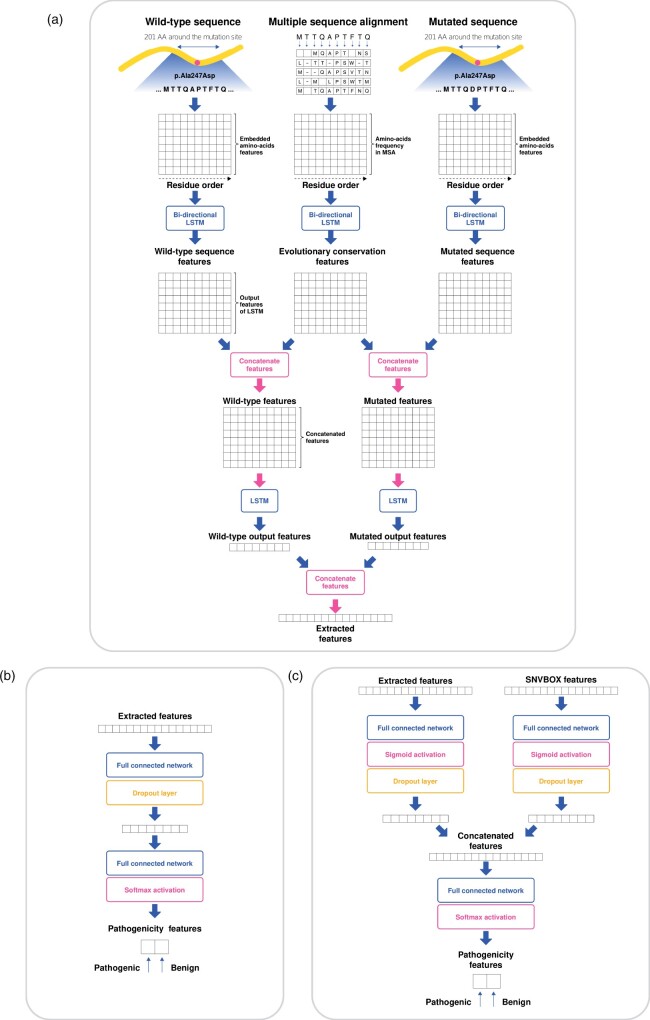
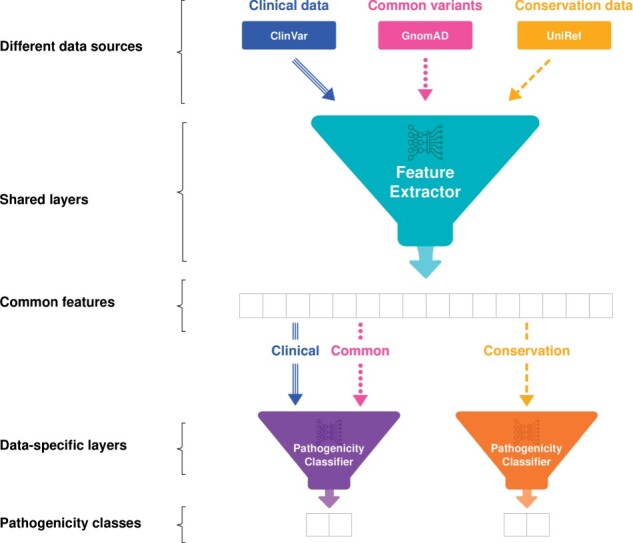
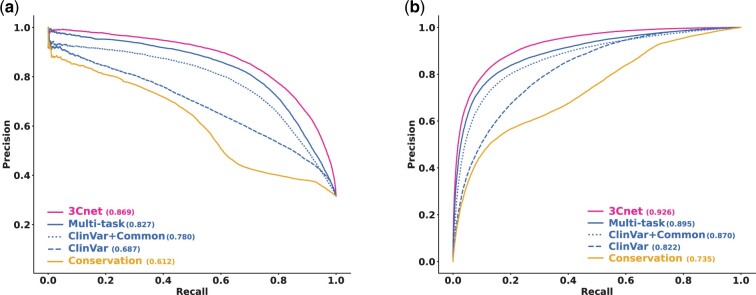
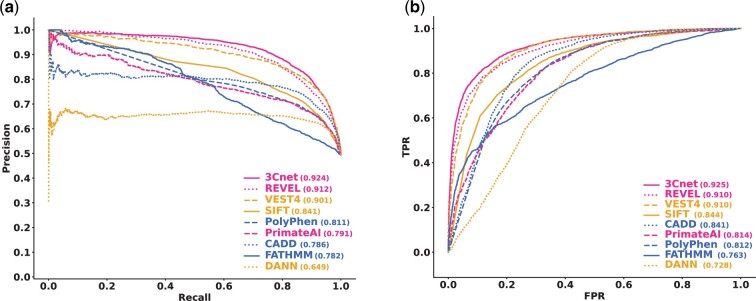
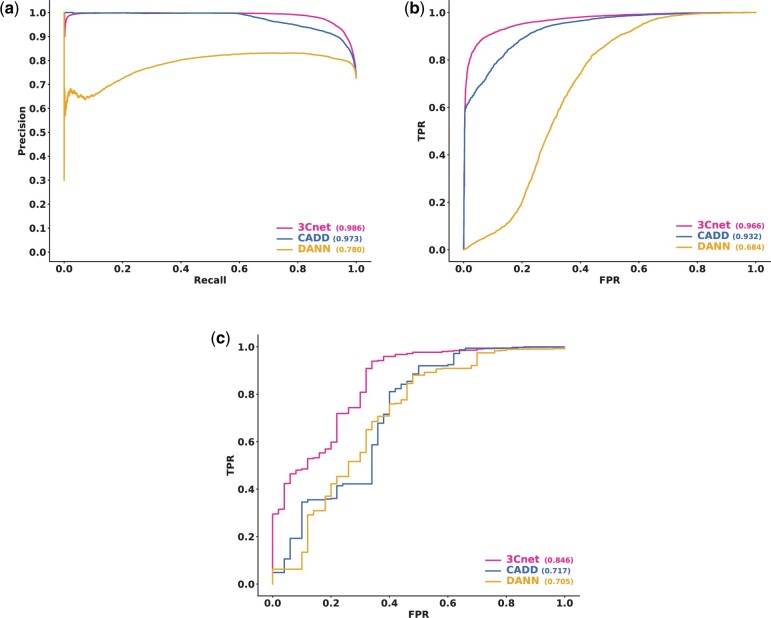
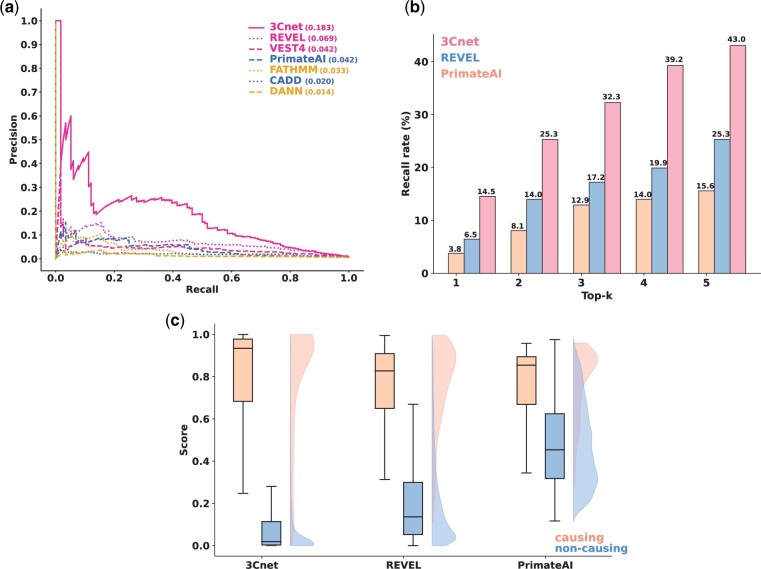
Results: We developed a pathogenicity predictor, 3Cnet, that uses recurrent neural networks to analyze the amino acid context of human variants. As 3Cnet is trained on simulated variants reflecting evolutionary conservation and clinical data, it can find disease-causing variants in patient genomes with 2.2 times greater sensitivity than currently available tools, more effectively discovering pathogenic variants and thereby improving diagnosis rates.
Availability and implementation: Codes (https://github.com/KyoungYeulLee/3Cnet/) and data (https://zenodo.org/record/4716879#.YIO-xqkzZH1) are freely available to non-commercial users.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press.
Figures
References
-
- Bleeker S.E. et al. (2003) External validation is necessary in prediction research: a clinical example. J. Clin. Epidemiol., 56, 826–832. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
