

An Update on Severe Acute Respiratory Syndrome Coronavirus 2 Diversity in the US National Capital Region: Evolution of Novel and Variants of Concern
- PMID: 34272947
- PMCID: PMC8406876
- DOI: 10.1093/cid/ciab636
An Update on Severe Acute Respiratory Syndrome Coronavirus 2 Diversity in the US National Capital Region: Evolution of Novel and Variants of Concern
Abstract
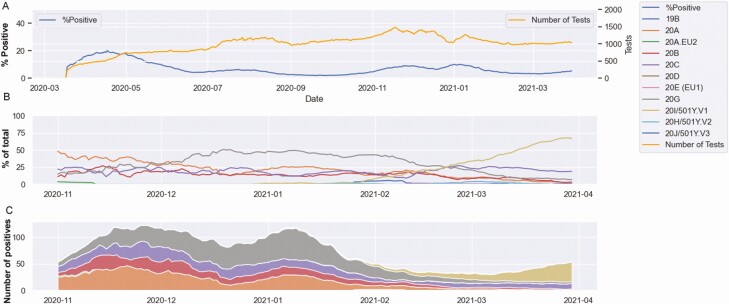
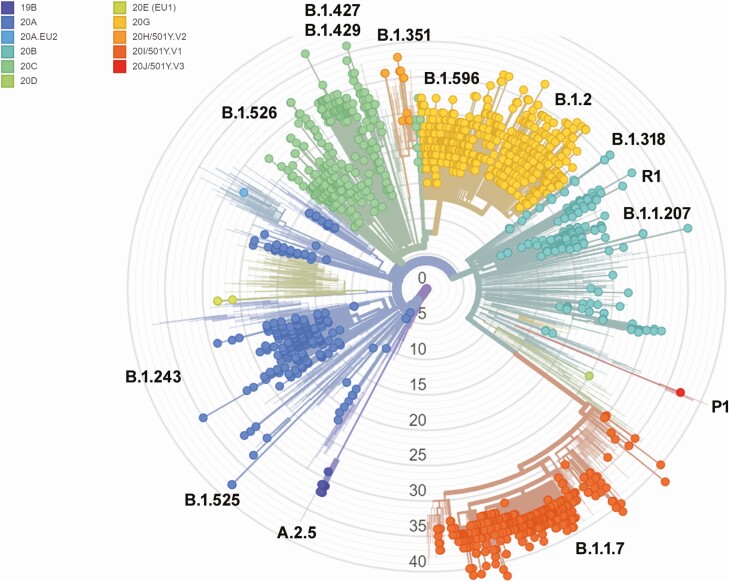
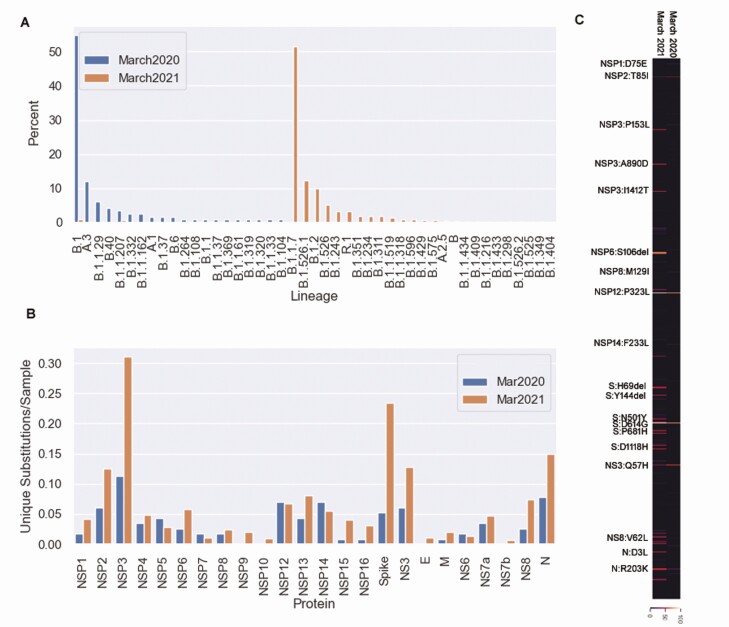
Background: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants concerning for enhanced transmission, evasion of immune responses, or associated with severe disease have motivated the global increase in genomic surveillance. In the current study, large-scale whole-genome sequencing was performed between November 2020 and the end of March 2021 to provide a phylodynamic analysis of circulating variants over time. In addition, we compared the viral genomic features of March 2020 and March 2021.
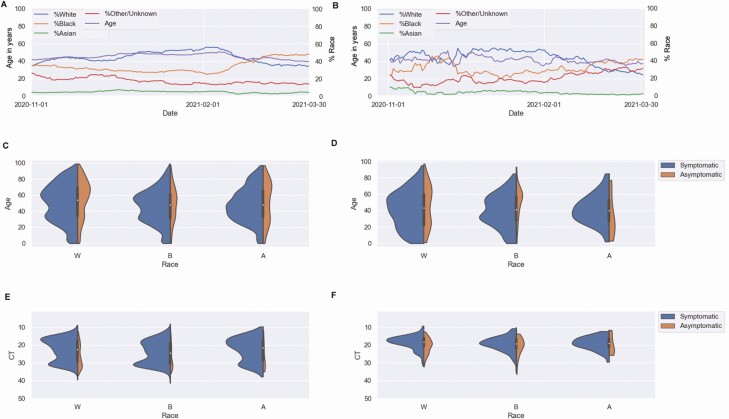
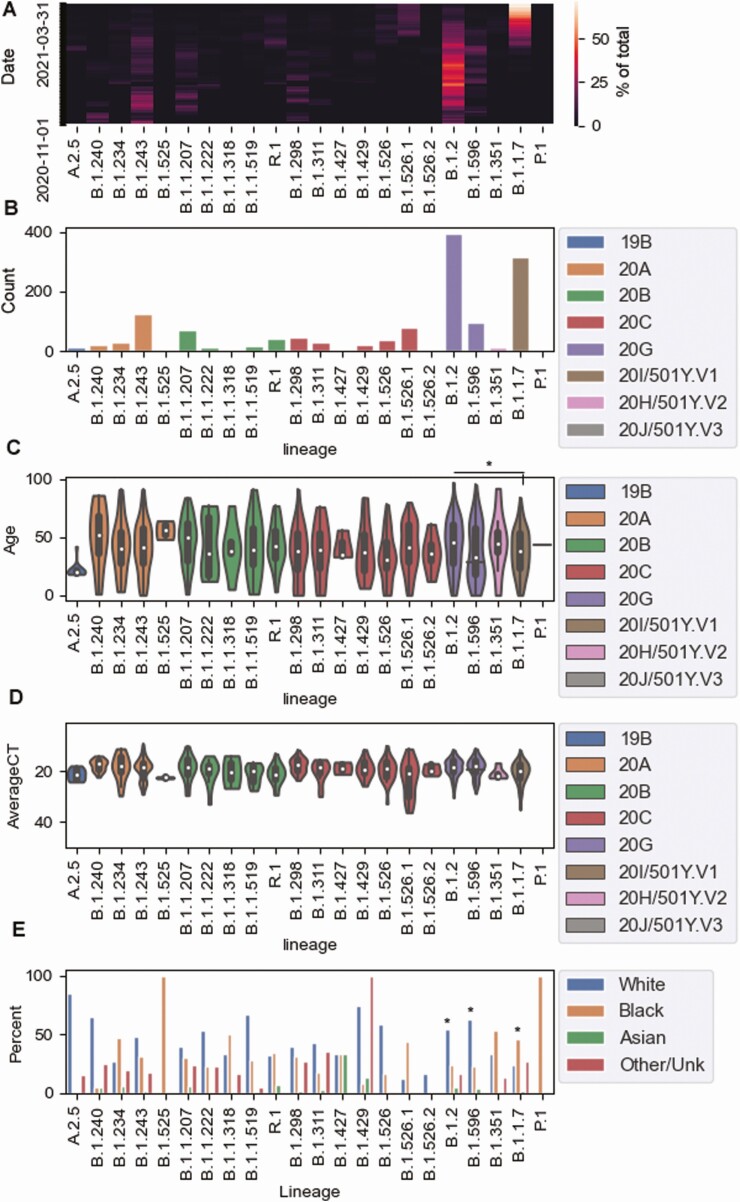
Methods: A total of 1600 complete SARS-CoV-2 genomes were analyzed. Genomic analysis was associated with laboratory diagnostic volumes and positivity rates, in addition to an analysis of the association of selected variants of concern/variants of interest with disease severity and outcomes. Our real-time surveillance features a cohort of specimens from patients who tested positive for SARS-CoV-2 after completion of vaccination.
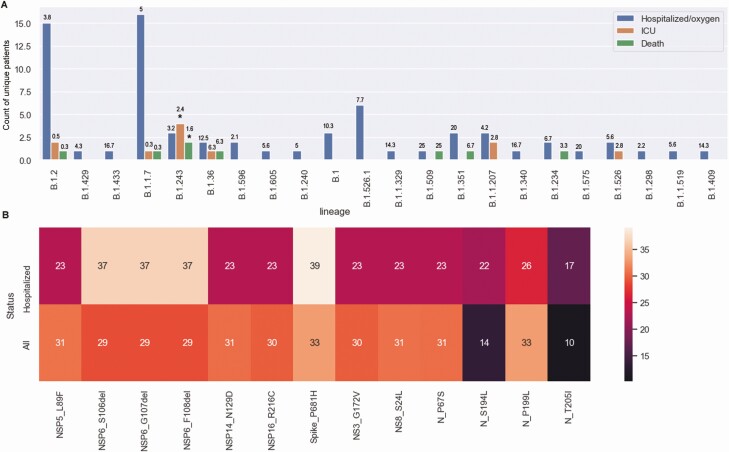
Results: Our data showed genomic diversity over time that was not limited to the spike sequence. A significant increase in the B.1.1.7 lineage (alpha variant) in March 2021 as well as a transient circulation of regional variants that carried both the concerning S: E484K and S: P681H substitutions were noted. Lineage B.1.243 was significantly associated with intensive care unit admission and mortality. Genomes recovered from fully vaccinated individuals represented the predominant lineages circulating at specimen collection time, and people with those infections recovered with no hospitalizations.
Conclusions: Our results emphasize the importance of genomic surveillance coupled with laboratory, clinical, and metadata analysis for a better understanding of the dynamics of viral spread and evolution.
Keywords: COVD-19; SARS-CoV-2; sequencing; variant of concern.
© The Author(s) 2021. Published by Oxford University Press for the Infectious Diseases Society of America. All rights reserved. For permissions, e-mail: journals.permissions@oup.com.
Figures
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
