

Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens
- PMID: 34278265
- PMCID: PMC8264160
- DOI: 10.1016/j.isci.2021.102757
Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens
Abstract
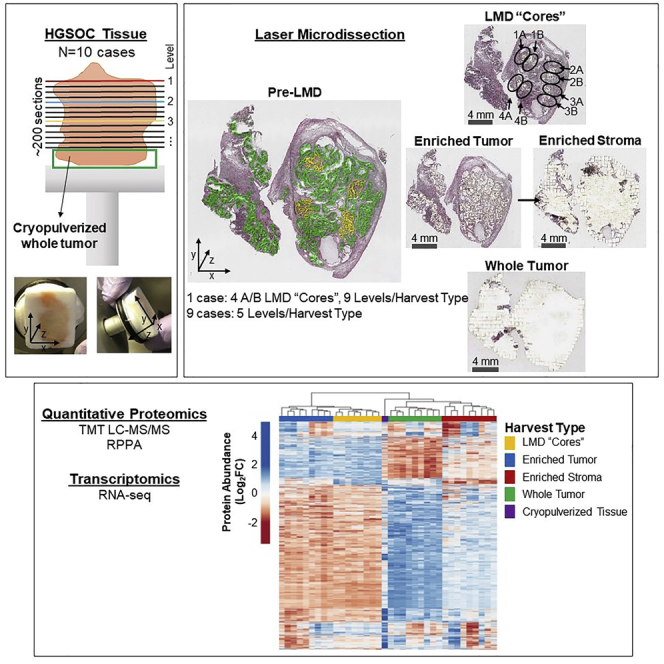
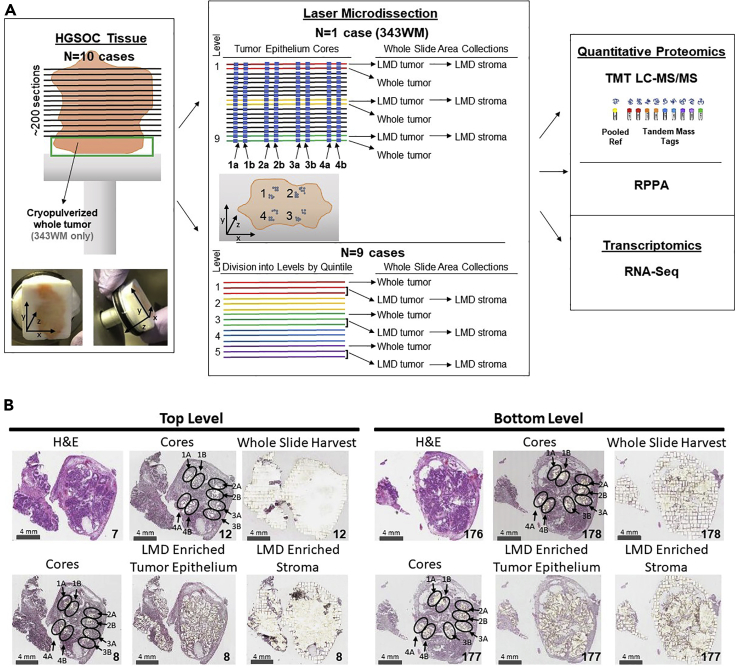
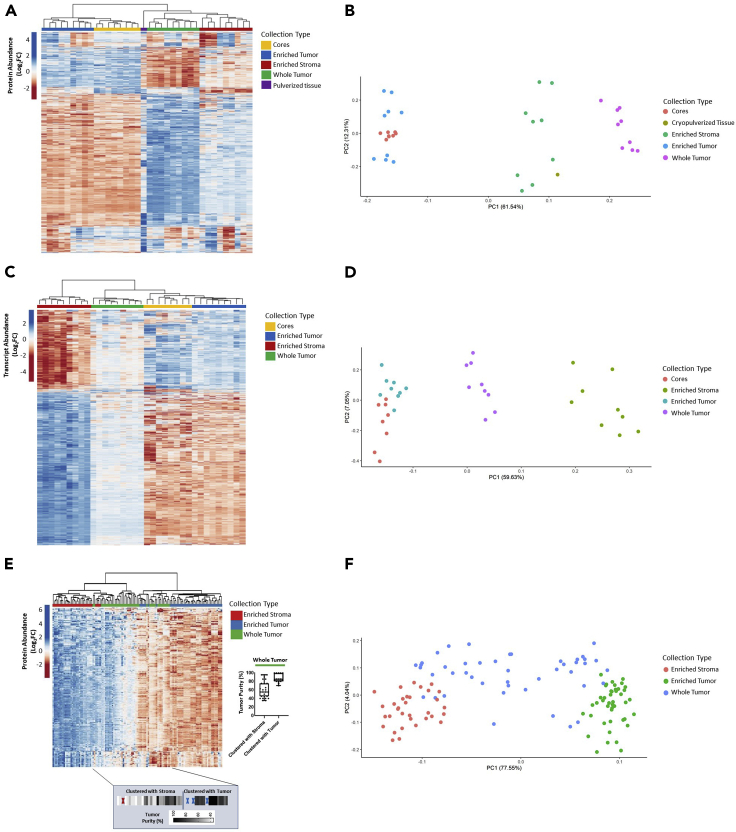
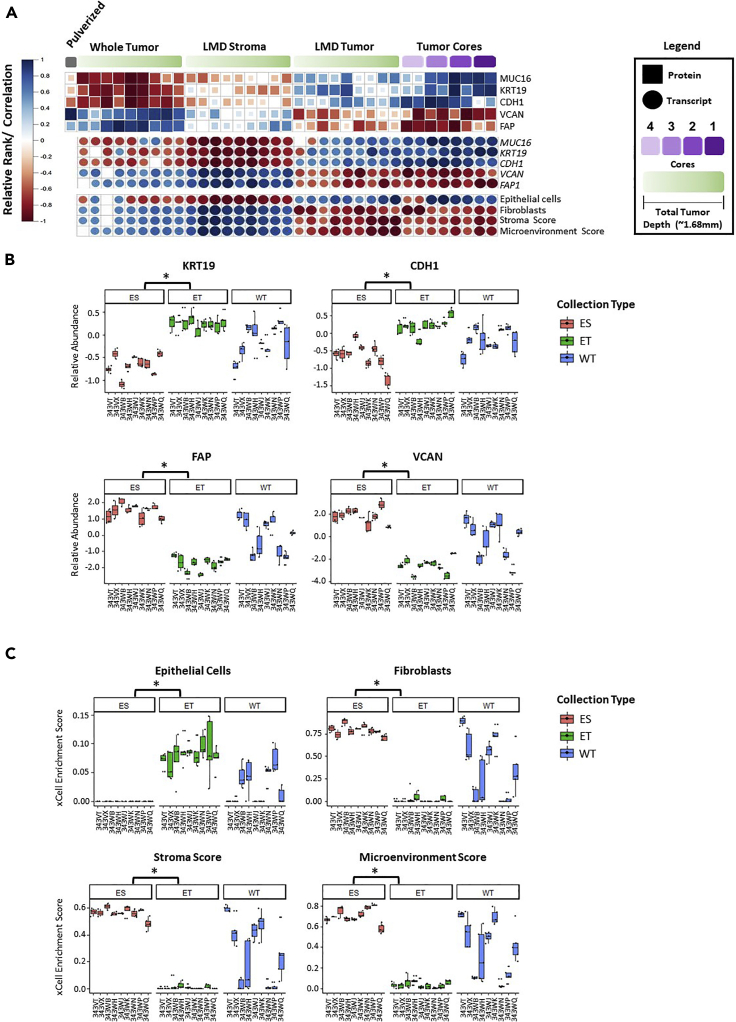
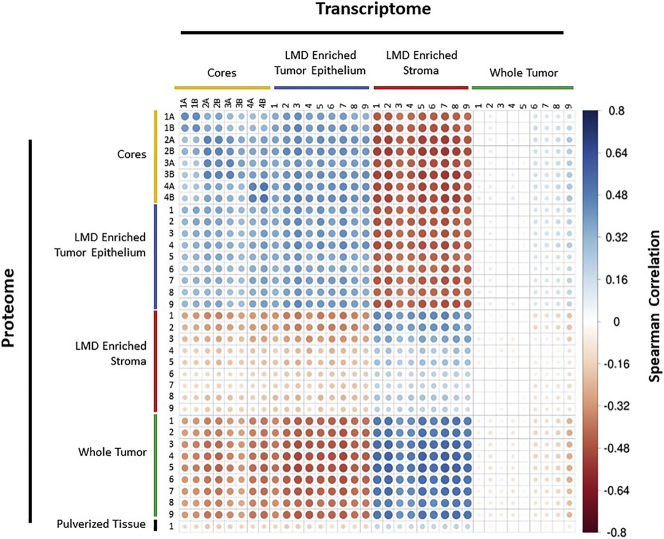
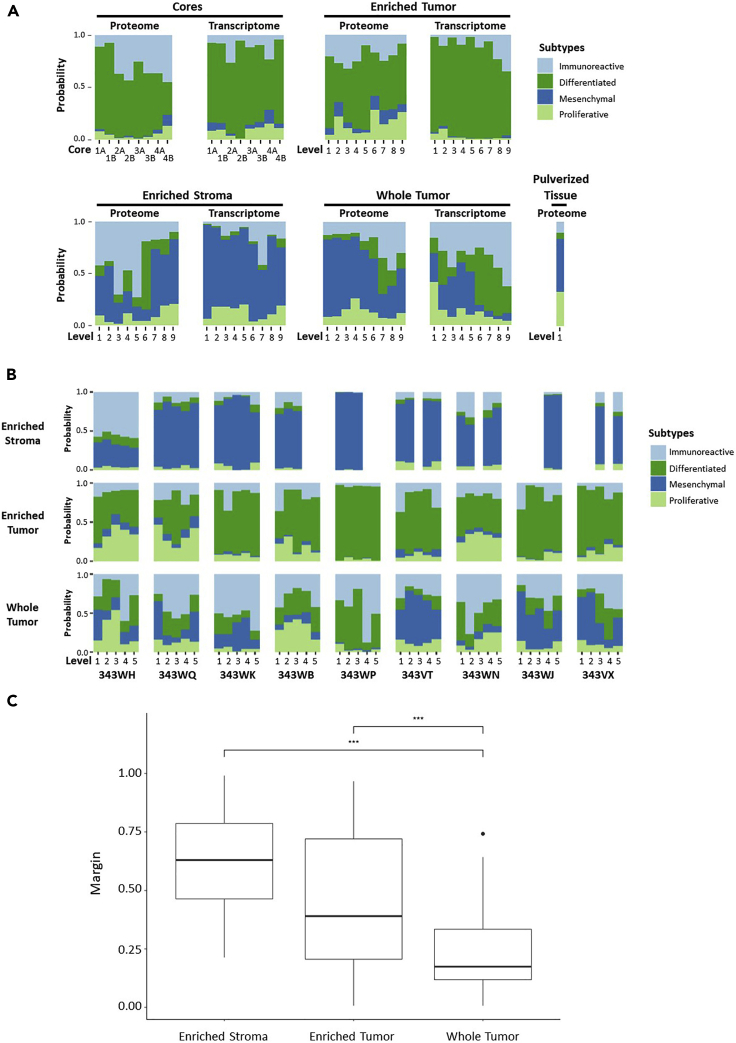
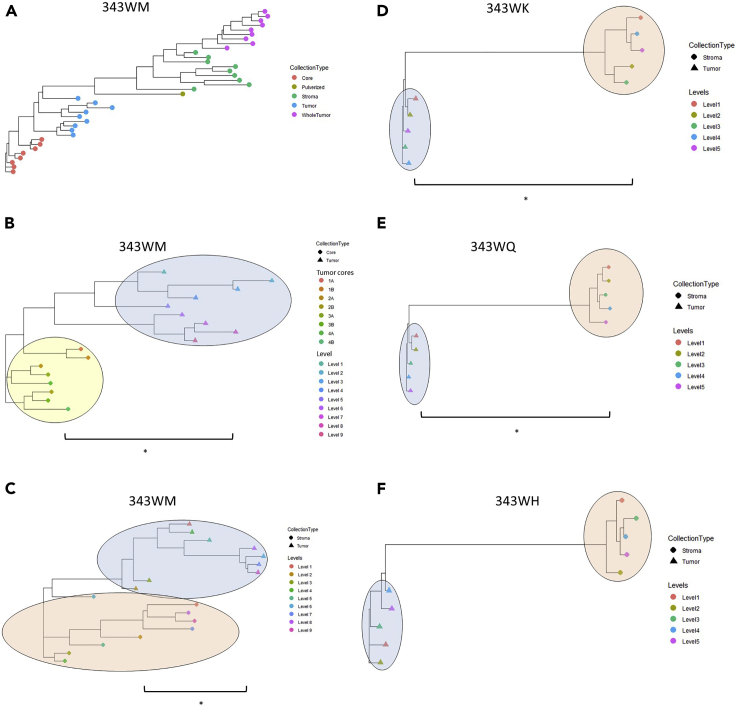
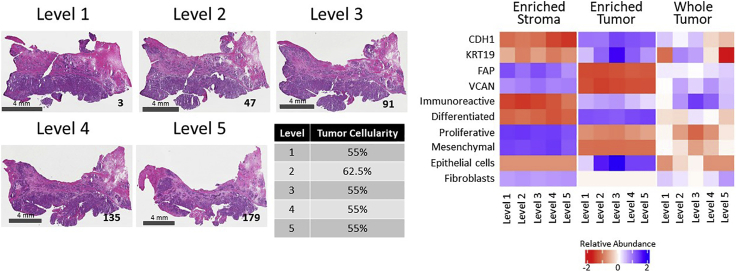
Enriched tumor epithelium, tumor-associated stroma, and whole tissue were collected by laser microdissection from thin sections across spatially separated levels of ten high-grade serous ovarian carcinomas (HGSOCs) and analyzed by mass spectrometry, reverse phase protein arrays, and RNA sequencing. Unsupervised analyses of protein abundance data revealed independent clustering of an enriched stroma and enriched tumor epithelium, with whole tumor tissue clustering driven by overall tumor "purity." Comparing these data to previously defined prognostic HGSOC molecular subtypes revealed protein and transcript expression from tumor epithelium correlated with the differentiated subtype, whereas stromal proteins (and transcripts) correlated with the mesenchymal subtype. Protein and transcript abundance in the tumor epithelium and stroma exhibited decreased correlation in samples collected just hundreds of microns apart. These data reveal substantial tumor microenvironment protein heterogeneity that directly bears on prognostic signatures, biomarker discovery, and cancer pathophysiology and underscore the need to enrich cellular subpopulations for expression profiling.
Keywords: Cancer systems biology; Oncology; Proteomics; Transcriptomics.
© 2021 The Author(s).
Conflict of interest statement
T.P.C. is a ThermoFisher Scientific, Inc SAB member and receives research funding from 10.13039/100006483AbbVie. E.F.P. receives research funding from 10.13039/100004328Genentech, 10.13039/100004319Pfizer, and 10.13039/100006483AbbVie and is a co-inventor of the RPPA technology described herein and receives royalties on the related license agreements.
Figures
References
-
- Baldelli E., Calvert V., Hodge A., Vanmeter A., Petricoin E.F., Pierobon M. Reverse phase protein microarrays. In: Espina V., editor. Molecular Profiling: Methods and Protocols. Springer; 2017. pp. 149–169. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
