

Highly contiguous assemblies of 101 drosophilid genomes
- PMID: 34279216
- PMCID: PMC8337076
- DOI: 10.7554/eLife.66405
Highly contiguous assemblies of 101 drosophilid genomes
Erratum in
-
Correction: Highly contiguous assemblies of 101 drosophilid genomes.Elife. 2022 Mar 18;11:e78579. doi: 10.7554/eLife.78579. Elife. 2022. PMID: 35302486 Free PMC article.
Abstract
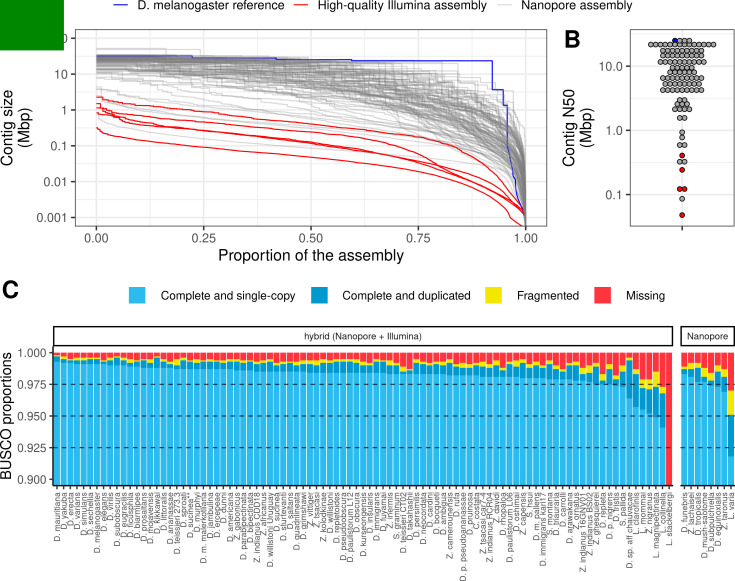
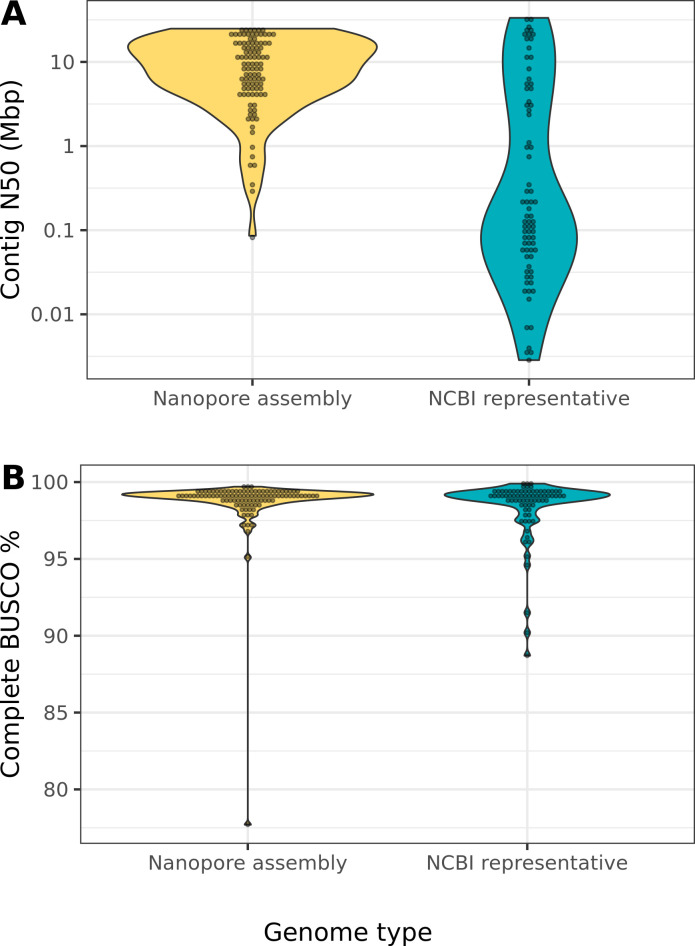
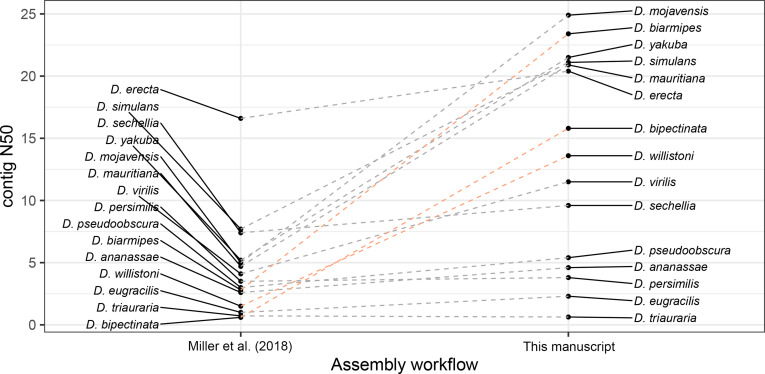
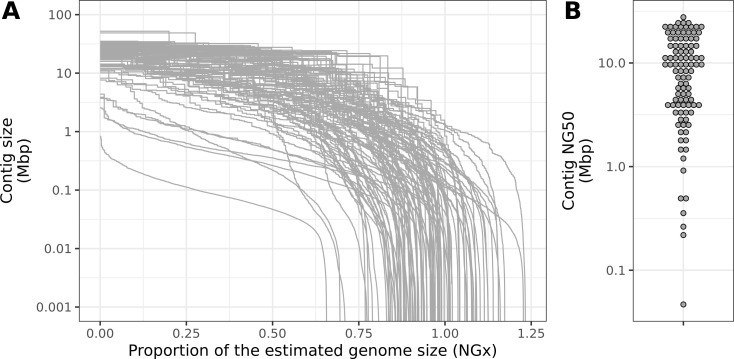
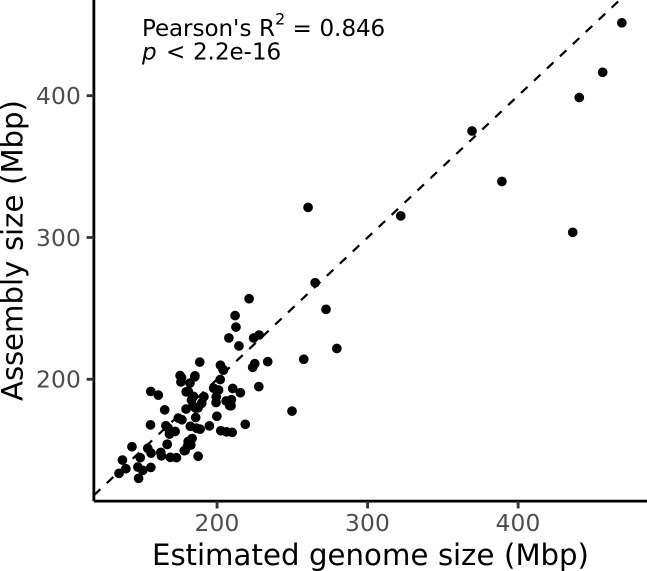
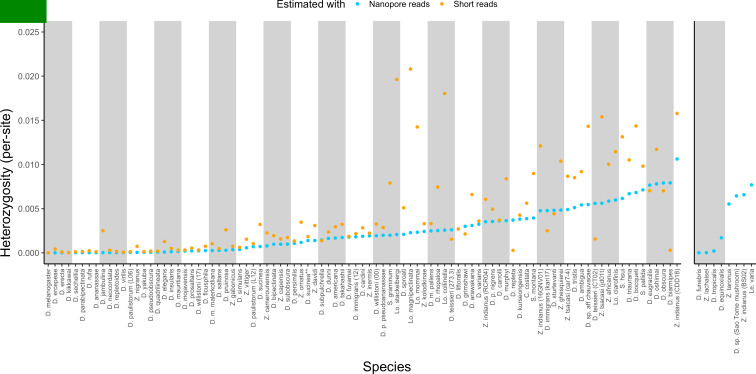
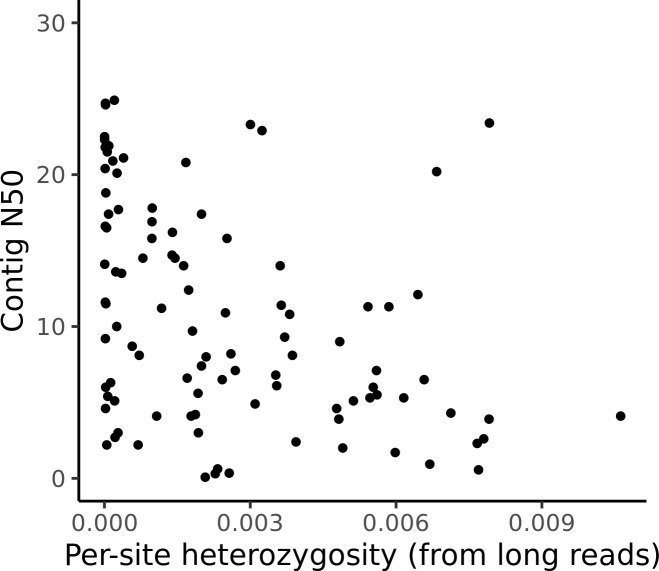
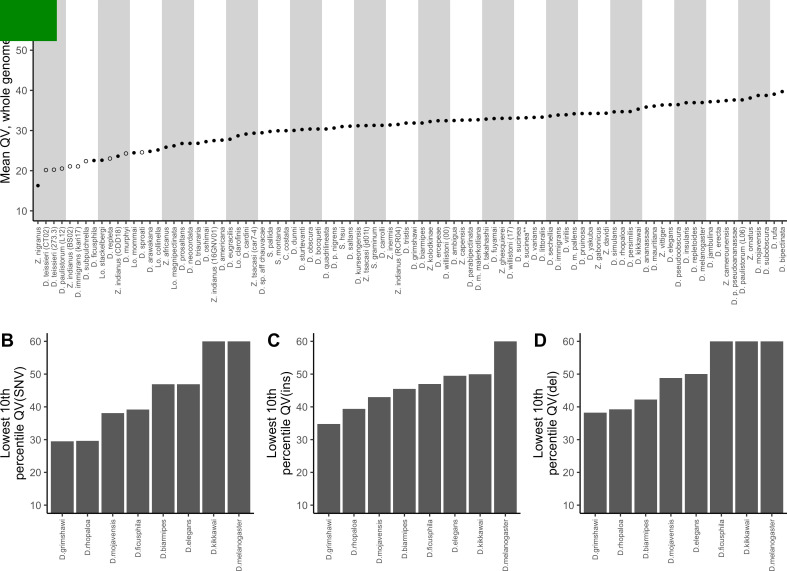
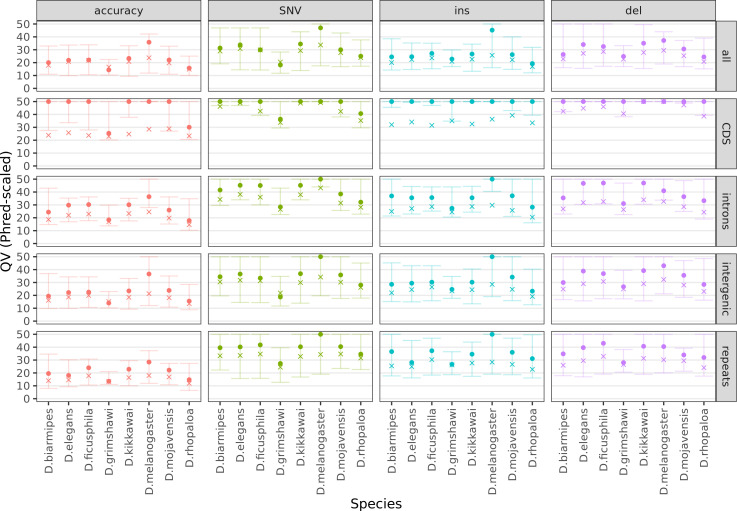
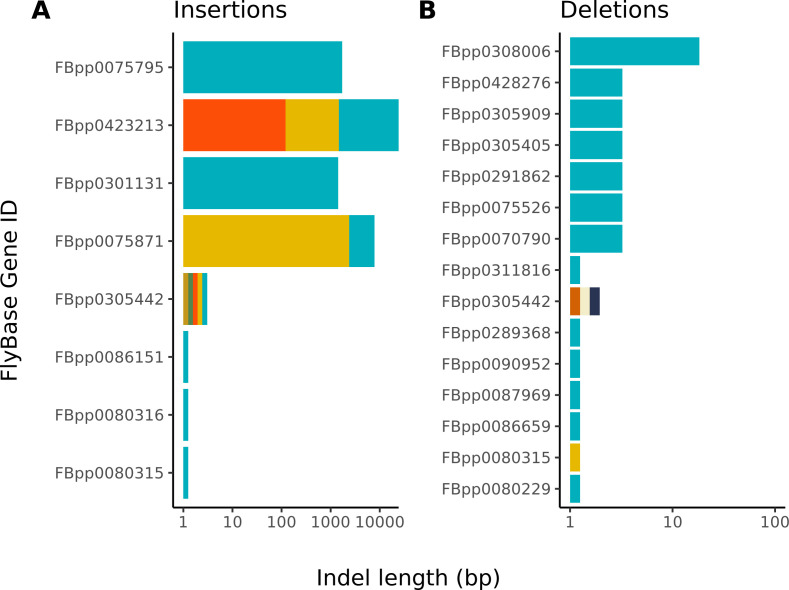
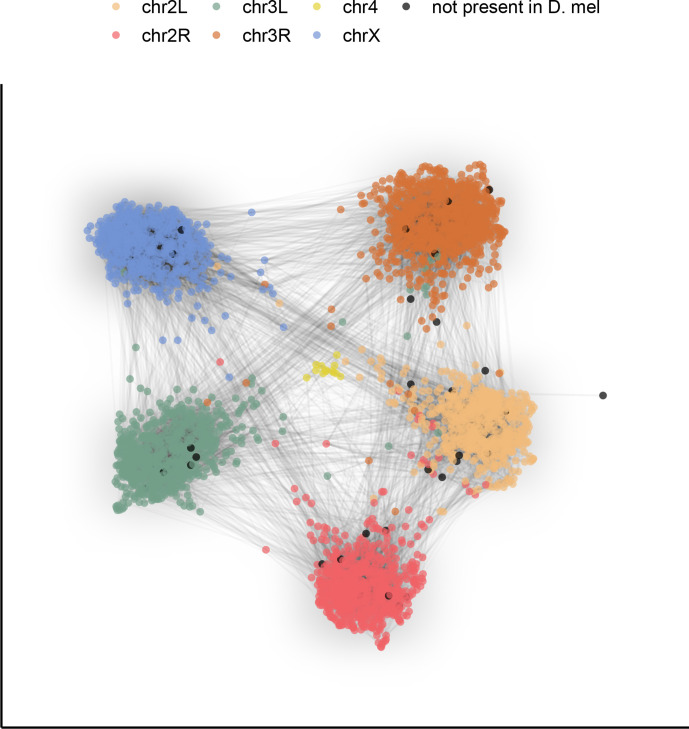
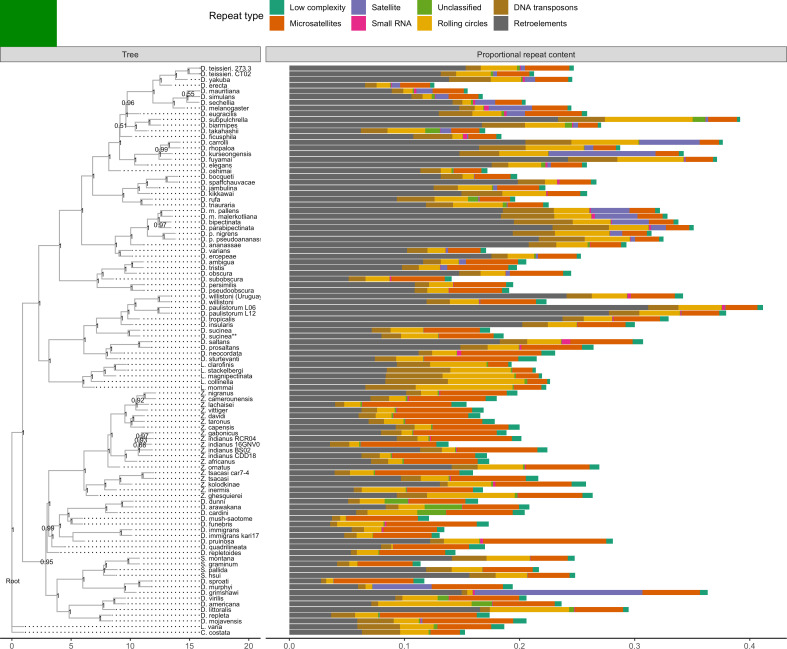
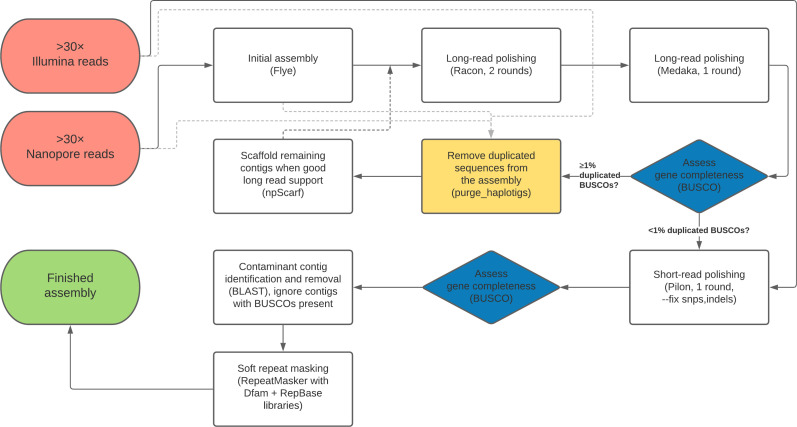
Over 100 years of studies in Drosophila melanogaster and related species in the genus Drosophila have facilitated key discoveries in genetics, genomics, and evolution. While high-quality genome assemblies exist for several species in this group, they only encompass a small fraction of the genus. Recent advances in long-read sequencing allow high-quality genome assemblies for tens or even hundreds of species to be efficiently generated. Here, we utilize Oxford Nanopore sequencing to build an open community resource of genome assemblies for 101 lines of 93 drosophilid species encompassing 14 species groups and 35 sub-groups. The genomes are highly contiguous and complete, with an average contig N50 of 10.5 Mb and greater than 97% BUSCO completeness in 97/101 assemblies. We show that Nanopore-based assemblies are highly accurate in coding regions, particularly with respect to coding insertions and deletions. These assemblies, along with a detailed laboratory protocol and assembly pipelines, are released as a public resource and will serve as a starting point for addressing broad questions of genetics, ecology, and evolution at the scale of hundreds of species.
Keywords: D. melanogaster; Drosophila; Drosophilidae; comparative genomics; evolutionary biology; genetics; genome assembly; genomics; long reads; nanopore.
© 2021, Kim et al.
Conflict of interest statement
BK, JW, DM, OB, ED, AT, AC, DP, ED, JP, JA, DH, TM, EA, MZ, YO, MS, MJ, MV, MT, PE, JG, TK, MT, HW, MW, JD, LM, GM, EB, VK, RH, AT, CJ, DP, NW, AK, DM, DP No competing interests declared
Figures
References
-
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RD, Scheeler F, Shen H, Shue BC, Sidén-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. - DOI - PubMed
-
- Armstrong J, Hickey G, Diekhans M, Fiddes IT, Novak AM, Deran A, Fang Q, Xie D, Feng S, Stiller J, Genereux D, Johnson J, Marinescu VD, Alföldi J, Harris RS, Lindblad-Toh K, Haussler D, Karlsson E, Jarvis ED, Zhang G, Paten B. Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature. 2020;587:246–251. doi: 10.1038/s41586-020-2871-y. - DOI - PMC - PubMed
-
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology : a journal of computational molecular cell biology. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
