

Epileptic Mechanisms Shared by Alzheimer's Disease: Viewed via the Unique Lens of Genetic Epilepsy
- PMID: 34281185
- PMCID: PMC8268161
- DOI: 10.3390/ijms22137133
Epileptic Mechanisms Shared by Alzheimer's Disease: Viewed via the Unique Lens of Genetic Epilepsy
Abstract
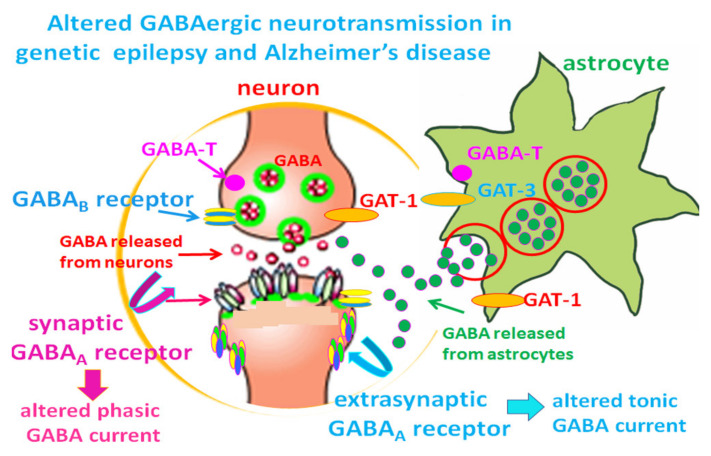
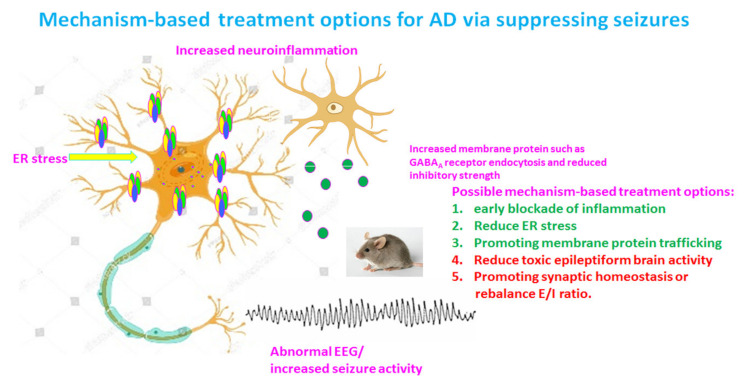
Our recent work on genetic epilepsy (GE) has identified common mechanisms between GE and neurodegenerative diseases including Alzheimer's disease (AD). Although both disorders are seemingly unrelated and occur at opposite ends of the age spectrum, it is likely there are shared mechanisms and studies on GE could provide unique insights into AD pathogenesis. Neurodegenerative diseases are typically late-onset disorders, but the underlying pathology may have already occurred long before the clinical symptoms emerge. Pathophysiology in the early phase of these diseases is understudied but critical for developing mechanism-based treatment. In AD, increased seizure susceptibility and silent epileptiform activity due to disrupted excitatory/inhibitory (E/I) balance has been identified much earlier than cognition deficit. Increased epileptiform activity is likely a main pathology in the early phase that directly contributes to impaired cognition. It is an enormous challenge to model the early phase of pathology with conventional AD mouse models due to the chronic disease course, let alone the complex interplay between subclinical nonconvulsive epileptiform activity, AD pathology, and cognition deficit. We have extensively studied GE, especially with gene mutations that affect the GABA pathway such as mutations in GABAA receptors and GABA transporter 1. We believe that some mouse models developed for studying GE and insights gained from GE could provide unique opportunity to understand AD. These include the pathology in early phase of AD, endoplasmic reticulum (ER) stress, and E/I imbalance as well as the contribution to cognitive deficit. In this review, we will focus on the overlapping mechanisms between GE and AD, the insights from mutations affecting GABAA receptors, and GABA transporter 1. We will detail mechanisms of E/I imbalance and the toxic epileptiform generation in AD, and the complex interplay between ER stress, impaired membrane protein trafficking, and synaptic physiology in both GE and AD.
Keywords: Alzheimer’s disease (AD); GABAergic signaling; amyloid β (Aβ); electroencephlography (EEG); endoplasmic reticulum (ER) stress; excitatory/inhibitory(E/I) imbalance; gamma-aminobutyric acid (GABA); genetic epilepsy (GE); protein trafficking.
Conflict of interest statement
The author declares no conflict of interest for this study.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
