

Amplicon and Metagenomic Analysis of Middle East Respiratory Syndrome (MERS) Coronavirus and the Microbiome in Patients with Severe MERS
- PMID: 34287009
- PMCID: PMC8386452
- DOI: 10.1128/mSphere.00219-21
Amplicon and Metagenomic Analysis of Middle East Respiratory Syndrome (MERS) Coronavirus and the Microbiome in Patients with Severe MERS
Abstract
Middle East respiratory syndrome coronavirus (MERS-CoV) is a zoonotic infection that emerged in the Middle East in 2012. Symptoms range from mild to severe and include both respiratory and gastrointestinal illnesses. The virus is mainly present in camel populations with occasional zoonotic spill over into humans. The severity of infection in humans is influenced by numerous factors, and similar to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), underlying health complications can play a major role. Currently, MERS-CoV and SARS-CoV-2 are coincident in the Middle East and thus a rapid way of sequencing MERS-CoV to derive genotype information for molecular epidemiology is needed. Additionally, complicating factors in MERS-CoV infections are coinfections that require clinical management. The ability to rapidly characterize these infections would be advantageous. To rapidly sequence MERS-CoV, an amplicon-based approach was developed and coupled to Oxford Nanopore long read length sequencing. This and a metagenomic approach were evaluated with clinical samples from patients with MERS. The data illustrated that whole-genome or near-whole-genome information on MERS-CoV could be rapidly obtained. This approach provided data on both consensus genomes and the presence of minor variants, including deletion mutants. The metagenomic analysis provided information of the background microbiome. The advantage of this approach is that insertions and deletions can be identified, which are the major drivers of genotype change in coronaviruses. IMPORTANCE Middle East respiratory syndrome coronavirus (MERS-CoV) emerged in late 2012 in Saudi Arabia. The virus is a serious threat to people not only in the Middle East but also in the world and has been detected in over 27 countries. MERS-CoV is spreading in the Middle East and neighboring countries, and approximately 35% of reported patients with this virus have died. This is the most severe coronavirus infection so far described. Saudi Arabia is a destination for many millions of people in the world who visit for religious purposes (Umrah and Hajj), and so it is a very vulnerable area, which imposes unique challenges for effective control of this epidemic. The significance of our study is that clinical samples from patients with MERS were used for rapid in-depth sequencing and metagenomic analysis using long read length sequencing.
Keywords: MERS-CoV; MinION; metagenomics; sequencing.
Figures

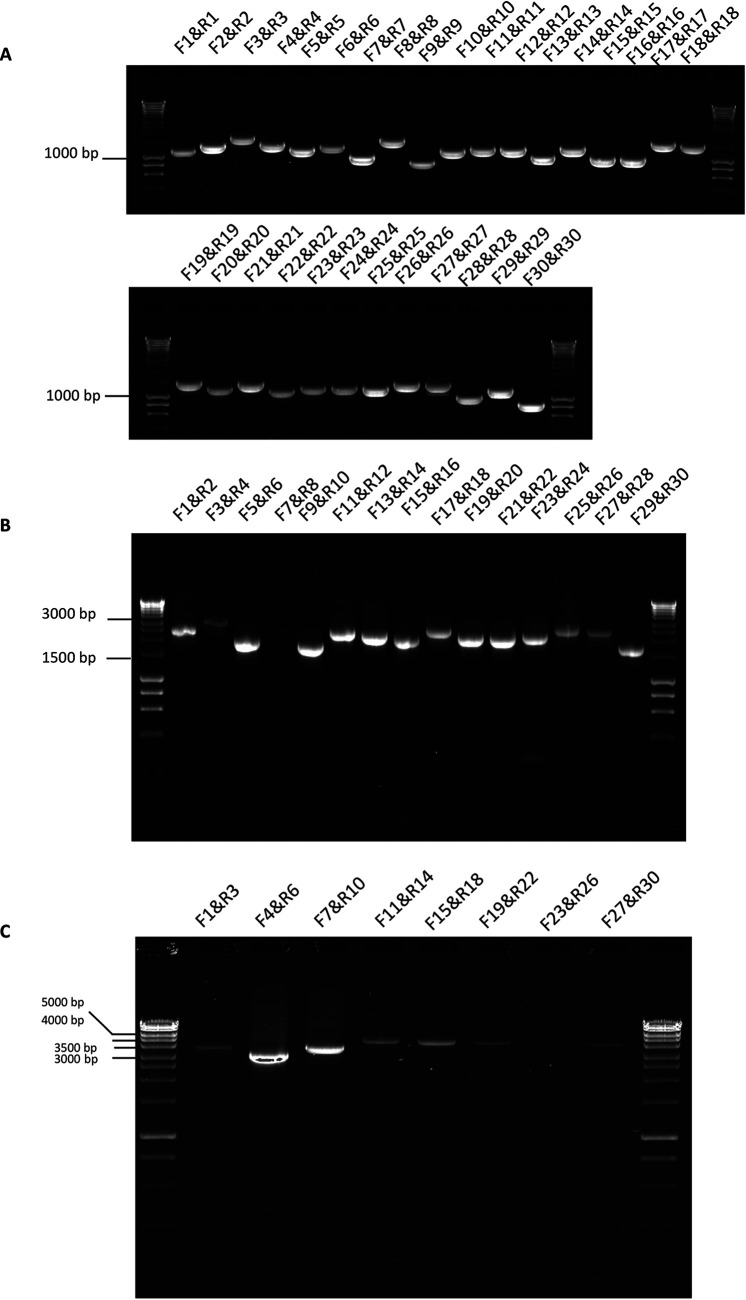
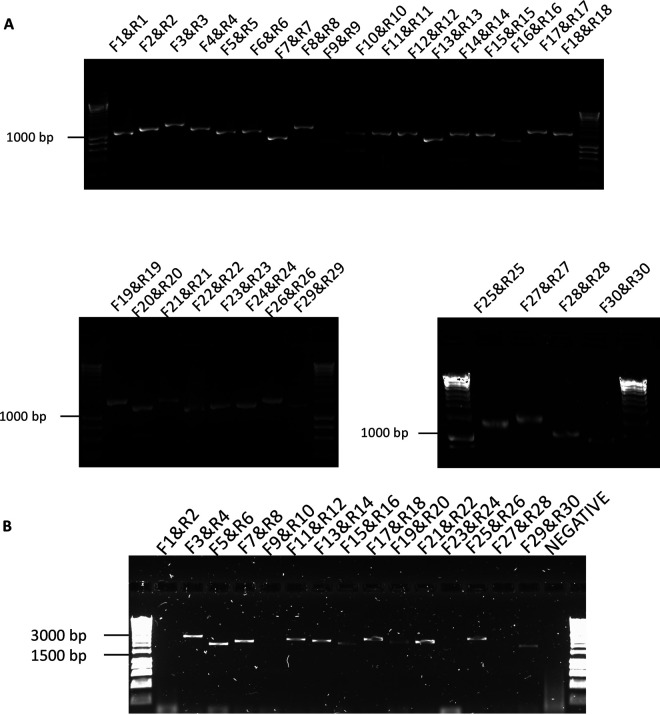
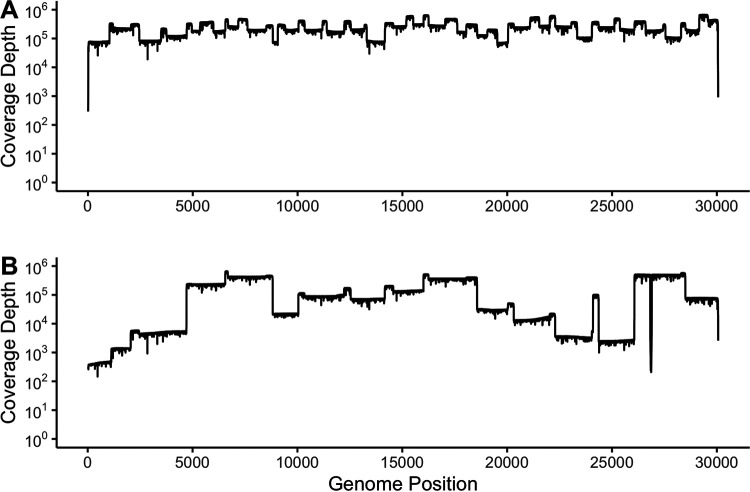
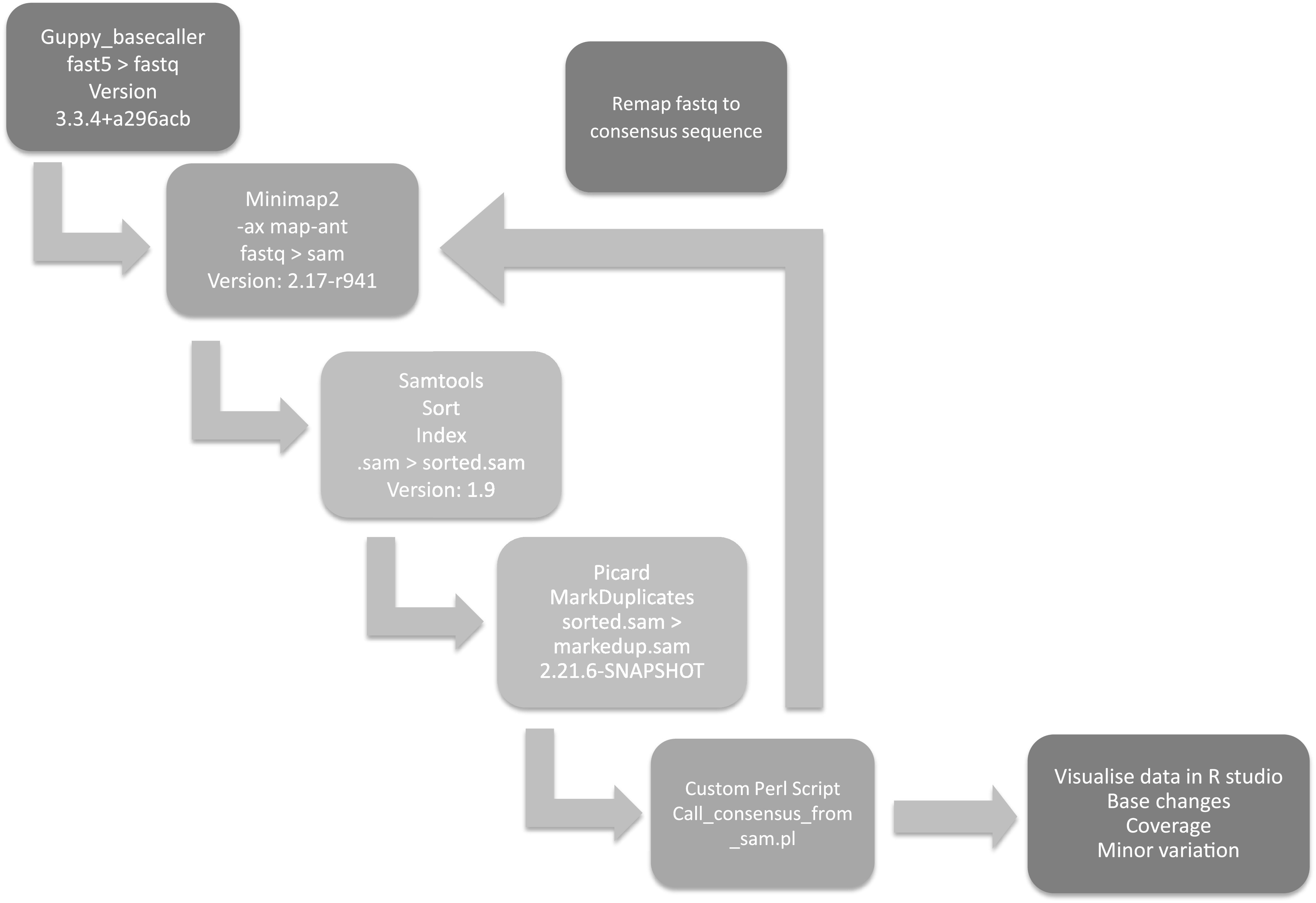
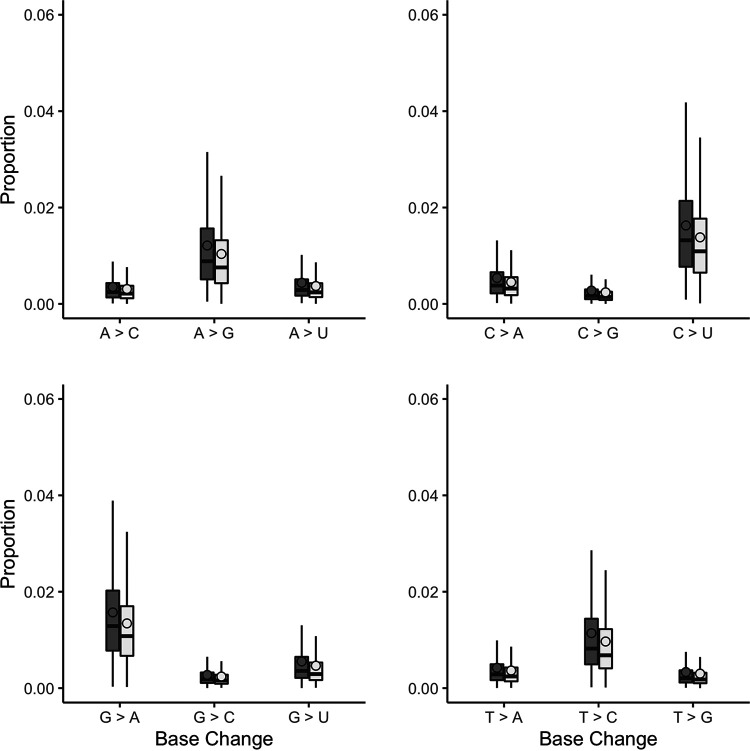
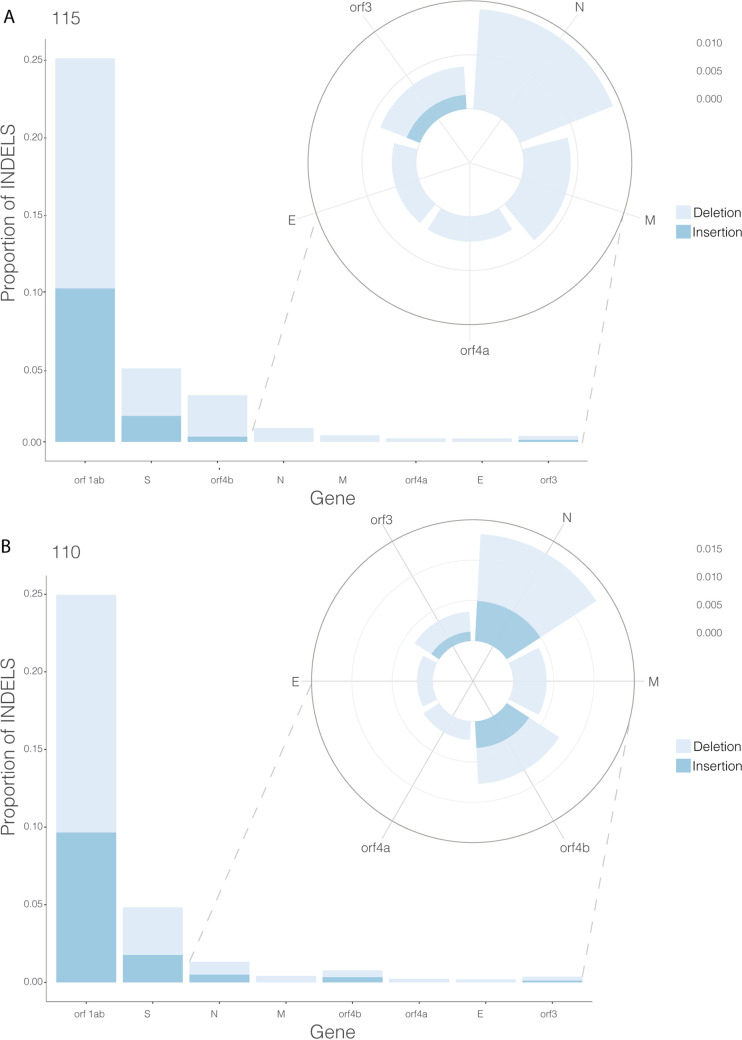
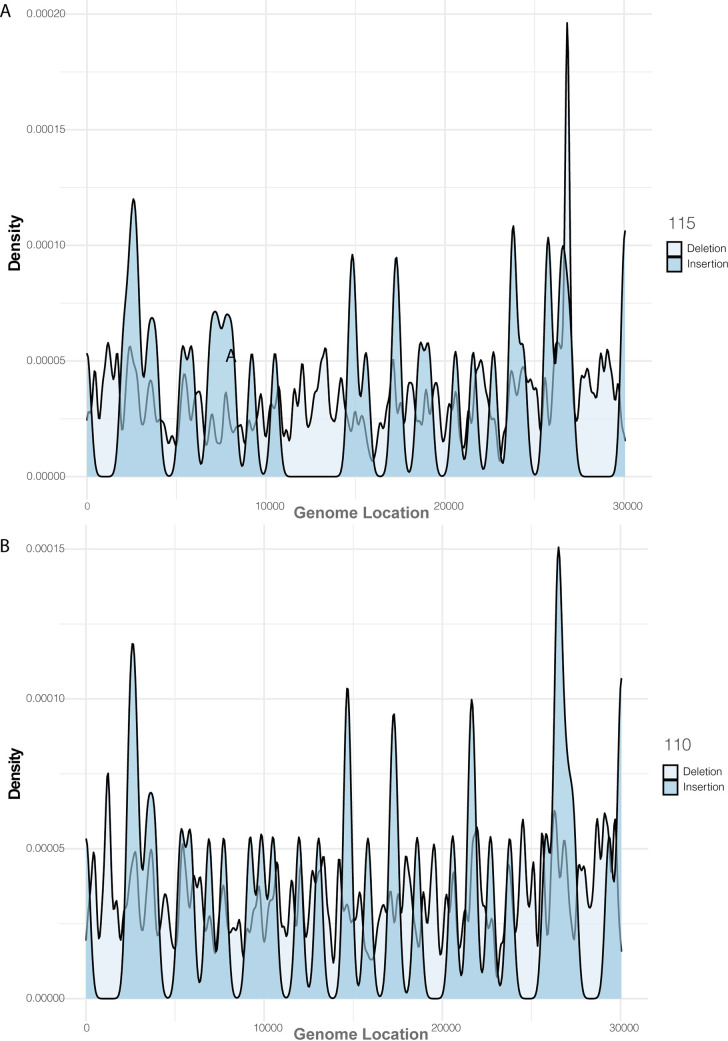
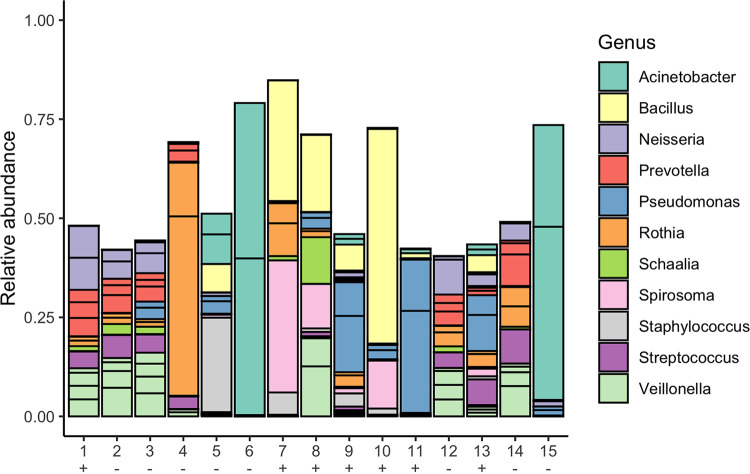
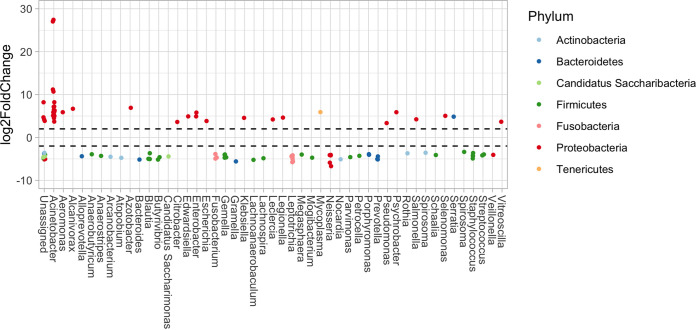
References
-
- Knight SR, Ho A, Pius R, Buchan I, Carson G, Drake TM, Dunning J, Fairfield CJ, Gamble C, Green CA, Gupta R, Halpin S, Hardwick HE, Holden KA, Horby PW, Jackson C, McLean KA, Merson L, Nguyen-Van-Tam JS, Norman L, Noursadeghi M, Olliaro PL, Pritchard MG, Russell CD, Shaw CA, Sheikh A, Solomon T, Sudlow C, Swann OV, Turtle LC, Openshaw PJ, Baillie JK, Semple MG, Docherty AB, Harrison EM, ISARIC4C Investigators . 2020. Risk stratification of patients admitted to hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: development and validation of the 4C Mortality Score. BMJ 370:m3339. doi: 10.1136/bmj.m3339. - DOI - PMC - PubMed
-
- Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, Walker S, Parkinson N, Fourman MH, Russell CD, Furniss J, Richmond A, Gountouna E, Wrobel N, Harrison D, Wang B, Wu Y, Meynert A, Griffiths F, Oosthuyzen W, Kousathanas A, Moutsianas L, Yang Z, Zhai R, Zheng C, Grimes G, Beale R, Millar J, Shih B, Keating S, Zechner M, Haley C, Porteous DJ, Hayward C, Yang J, Knight J, Summers C, Shankar-Hari M, Klenerman P, Turtle L, Ho A, Moore SC, Hinds C, Horby P, Nichol A, Maslove D, Ling L, McAuley D, Montgomery H, Walsh T, Gen-COVID Investigators , et al. 2021. Genetic mechanisms of critical illness in COVID-19. Nature 591:92–98. doi: 10.1038/s41586-020-03065-y. - DOI - PubMed
-
- Drake TM, Docherty AB, Harrison EM, Quint JK, Adamali H, Agnew S, Babu S, Barber CM, Barratt S, Bendstrup E, Bianchi S, Villegas DC, Chaudhuri N, Chua F, Coker R, Chang W, Crawshaw A, Crowley LE, Dosanjh D, Fiddler CA, Forrest IA, George PM, Gibbons MA, Groom K, Haney S, Hart SP, Heiden E, Henry M, Ho L-P, Hoyles RK, Hutchinson J, Hurley K, Jones M, Jones S, Kokosi M, Kreuter M, MacKay LS, Mahendran S, Margaritopoulos G, Molina-Molina M, Molyneaux PL, O’Brien A, O’Reilly K, Packham A, Parfrey H, Poletti V, Porter JC, Renzoni E, Rivera-Ortega P, Russell A-M, et al. 2020. Outcome of hospitalization for COVID-19 in patients with interstitial lung disease. An international multicenter study. Am J Respir Crit Care Med 202:1656–1665. doi: 10.1164/rccm.202007-2794OC. - DOI - PMC - PubMed
-
- Dorward DA, Russell CD, Um IH, Elshani M, Armstrong SD, Penrice-Randal R, Millar T, Lerpiniere CEB, Tagliavini G, Hartley CS, Randle NP, Gachanja NN, Potey PMD, Dong X, Anderson AM, Campbell VL, Duguid AJ, Al Qsous W, BouHaidar R, Baillie JK, Dhaliwal K, Wallace WA, Bellamy COC, Prost S, Smith C, Hiscox JA, Harrison DJ, Lucas CD. 2021. Tissue-specific immunopathology in fatal COVID-19. Am J Respir Crit Care Med 203:192–201. doi: 10.1164/rccm.202008-3265OC. - DOI - PMC - PubMed
-
- Mostafa HH, Fissel JA, Fanelli B, Bergman Y, Gniazdowski V, Dadlani M, Carroll KC, Colwell RR, Simner PJ. 2020. Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect COVID-19 patients. mBio 11:e01969-20. doi: 10.1128/mBio.01969-20. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
