

Reversal of emphysema by restoration of pulmonary endothelial cells
- PMID: 34287647
- PMCID: PMC8298104
- DOI: 10.1084/jem.20200938
Reversal of emphysema by restoration of pulmonary endothelial cells
Abstract
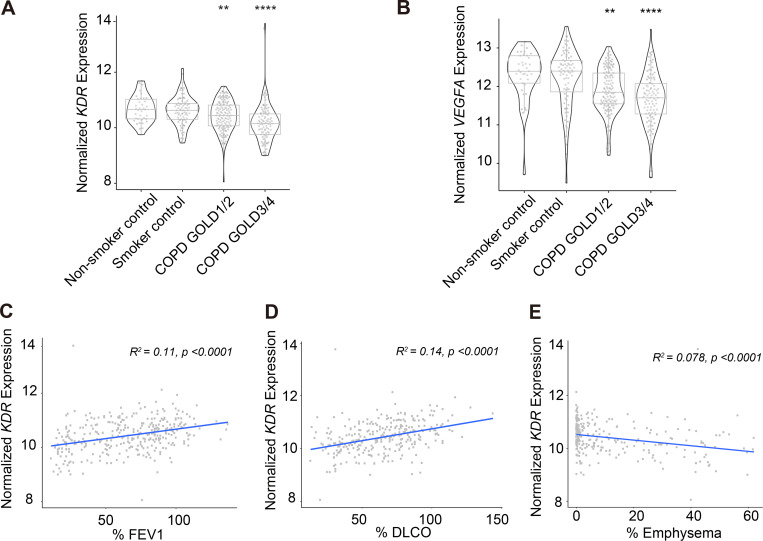
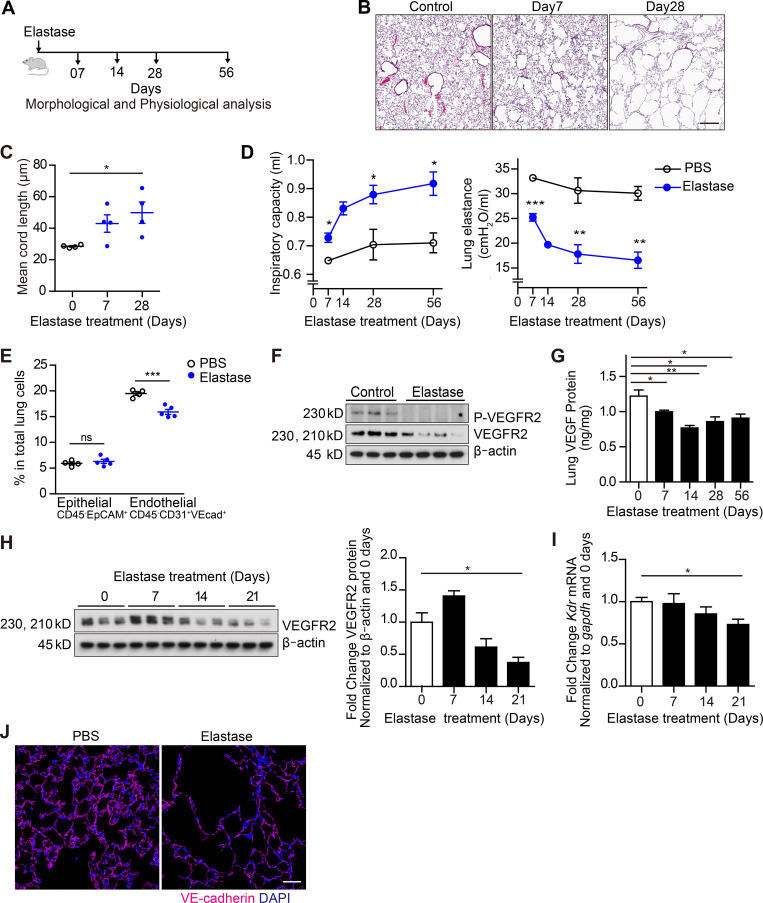
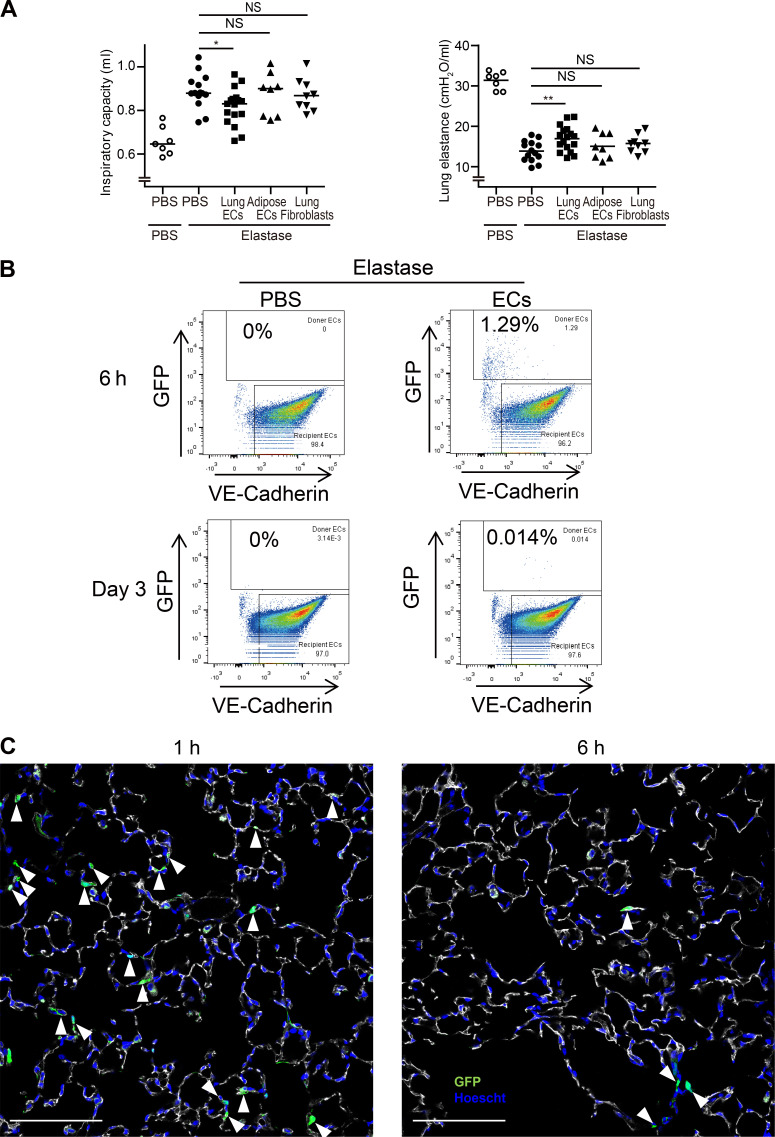
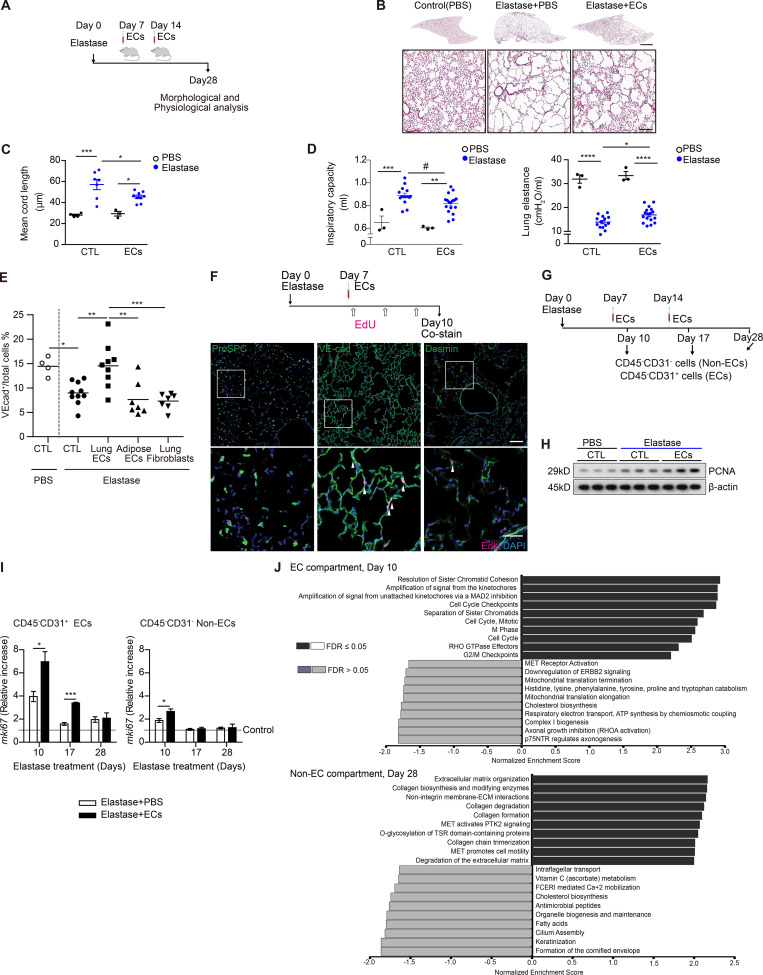
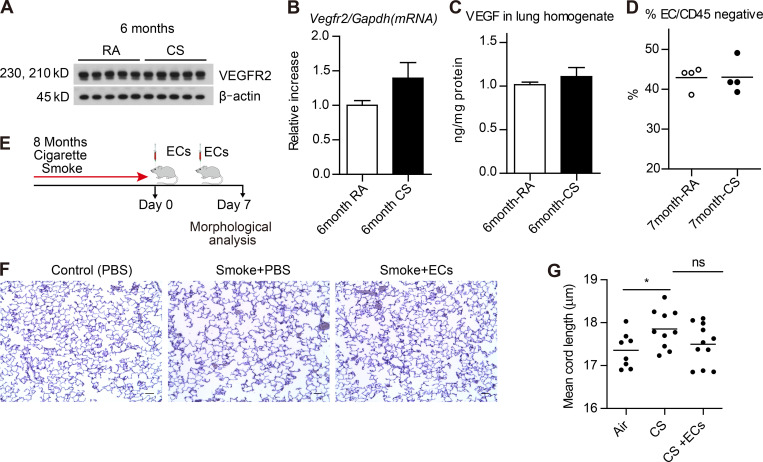
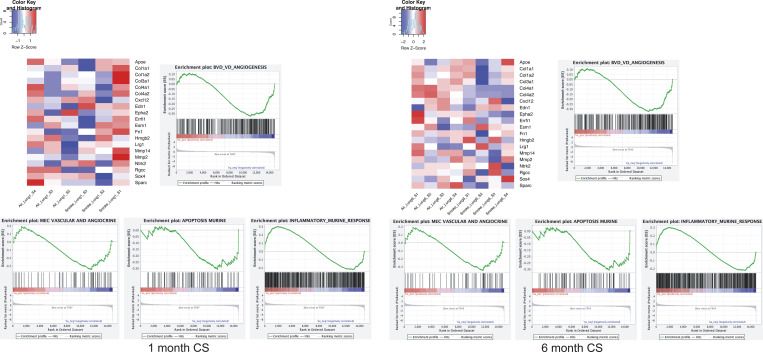
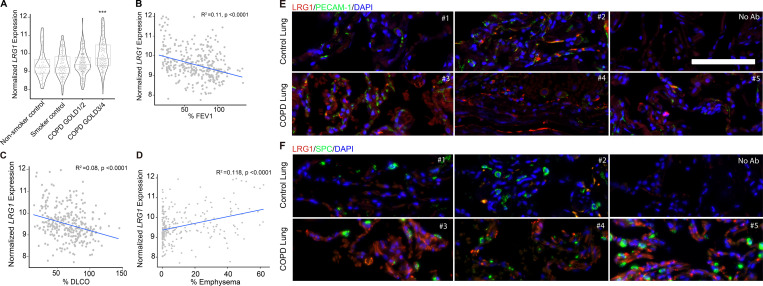
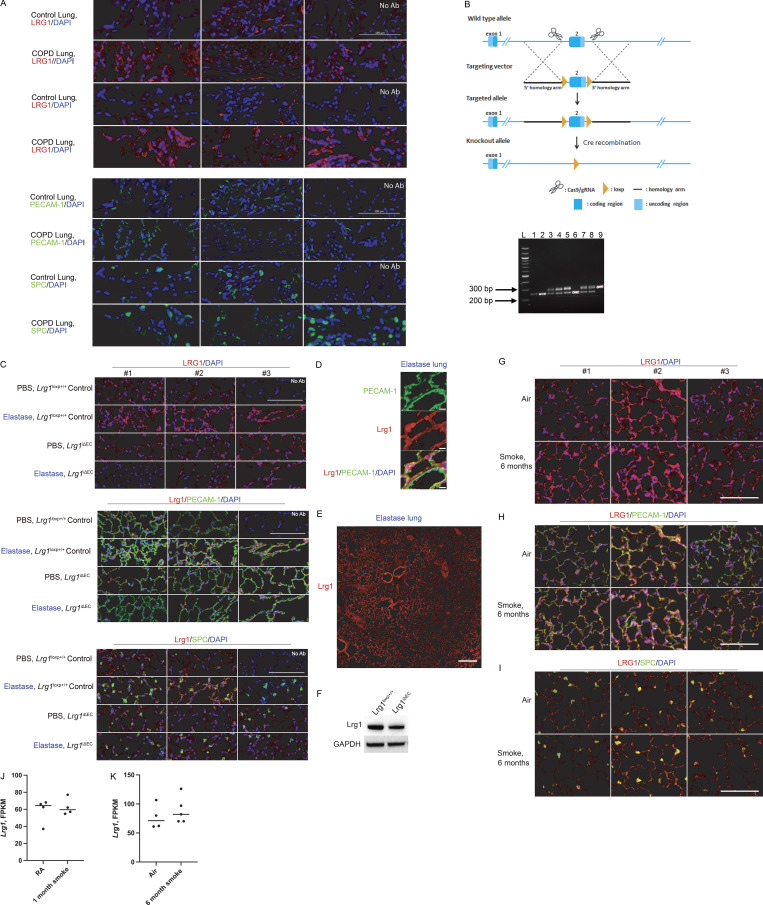
Chronic obstructive pulmonary disease (COPD) is marked by airway inflammation and airspace enlargement (emphysema) leading to airflow obstruction and eventual respiratory failure. Microvasculature dysfunction is associated with COPD/emphysema. However, it is not known if abnormal endothelium drives COPD/emphysema pathology and/or if correcting endothelial dysfunction has therapeutic potential. Here, we show the centrality of endothelial cells to the pathogenesis of COPD/emphysema in human tissue and using an elastase-induced murine model of emphysema. Airspace disease showed significant endothelial cell loss, and transcriptional profiling suggested an apoptotic, angiogenic, and inflammatory state. This alveolar destruction was rescued by intravenous delivery of healthy lung endothelial cells. Leucine-rich α-2-glycoprotein-1 (LRG1) was a driver of emphysema, and deletion of Lrg1 from endothelial cells rescued vascular rarefaction and alveolar regression. Hence, targeting endothelial cell biology through regenerative methods and/or inhibition of the LRG1 pathway may represent strategies of immense potential for the treatment of COPD/emphysema.
© 2021 Hisata et al.
Conflict of interest statement
Disclosures: D.J. Nolan reported personal fees from Angiocrine Bioscience outside the submitted work; in addition, D.J. Nolan had a patent number 8,465,732 issued (Angiocrine Bioscience) and a patent number 9,944,897 issued; and is an employee and equity holder of Angiocrine Bioscience. M. Ginsberg reported personal fees from Angiocrine Bioscience outside the submitted work; in addition, M. Ginsberg had a patent to 8,465,732 issued and a patent to 9,944,897 issued. In addition, M. Ginsberg is a current employee and equity holder of Angiocrine Bioscience. F.J. Martinez reported non-financial support from ProterrixBio, Nitto, Zambon; "other" from Afferent/Merck, Biogen, Veracyte, Prometic, Bridge Biotherapeutics, and Abbvie; grants from Gilead; and personal fees from AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Chiesi, Sunovion, Patara/Respivant, Bayer, Promedior/Roche, Teva, Col Behring, DevPro, IQVIA, Sanofi/Regeneron, United Therapeutics, and Novartis outside the submitted work. S.M. Cloonan reported grants from National Institute of Health, National Heart, Blood and Lung Institute (NHLBI), and Science Foundation Ireland (SFI), and personal fees from Pharmacosmos outside the submitted work; in addition, S.M. Cloonan had a patent number 10,905,682 issued. S. Rafii reported non-financial support from Angiocrine Bioscience during the conduct of the study; non-financial support from Angiocrine Bioscience outside the submitted work; and had a patent to E4ORF1 Endothelial cell infusion for organ repair licensed (Angiocrine Bioscience). A.M.K. Choi is a cofounder and equity stock holder for Proterris, which develops therapeutic uses for carbon monoxide. A.M.K. Choi has a use patent on CO. Additionally, A.M.K. Choi has a patent in COPD. No other disclosures were reported.
Figures




References
-
- Andersen, J.D., Boylan K.L., Jemmerson R., Geller M.A., Misemer B., Harrington K.M., Weivoda S., Witthuhn B.A., Argenta P., Vogel R.I., and Skubitz A.P.. 2010. Leucine-rich alpha-2-glycoprotein-1 is upregulated in sera and tumors of ovarian cancer patients. J. Ovarian Res. 3:21. 10.1186/1757-2215-3-21 - DOI - PMC - PubMed
-
- Barr, R.G., Mesia-Vela S., Austin J.H., Basner R.C., Keller B.M., Reeves A.P., Shimbo D., and Stevenson L.. 2007. Impaired flow-mediated dilation is associated with low pulmonary function and emphysema in ex-smokers: the Emphysema and Cancer Action Project (EMCAP) Study. Am. J. Respir. Crit. Care Med. 176:1200–1207. 10.1164/rccm.200707-980OC - DOI - PMC - PubMed
-
- Butler, J.M., Nolan D.J., Vertes E.L., Varnum-Finney B., Kobayashi H., Hooper A.T., Seandel M., Shido K., White I.A., Kobayashi M., et al. . 2010. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 6:251–264. 10.1016/j.stem.2010.02.001 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
