

Dynamic Expression of Transient Receptor Potential Vanilloid-3 and Integrated Signaling with Growth Factor Pathways during Lung Epithelial Wound Repair following Wood Smoke Particle and Other Forms of Lung Cell Injury
- PMID: 34290137
- PMCID: PMC11037451
- DOI: 10.1124/molpharm.121.000280
Dynamic Expression of Transient Receptor Potential Vanilloid-3 and Integrated Signaling with Growth Factor Pathways during Lung Epithelial Wound Repair following Wood Smoke Particle and Other Forms of Lung Cell Injury
Abstract
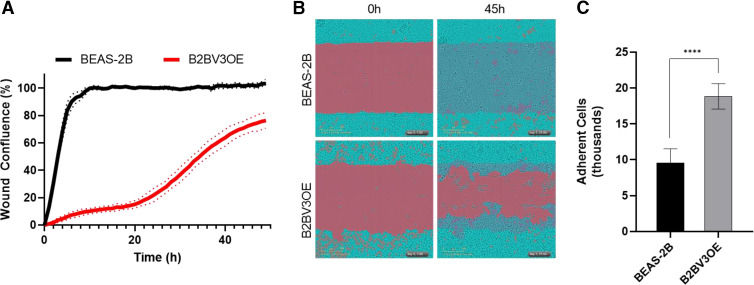
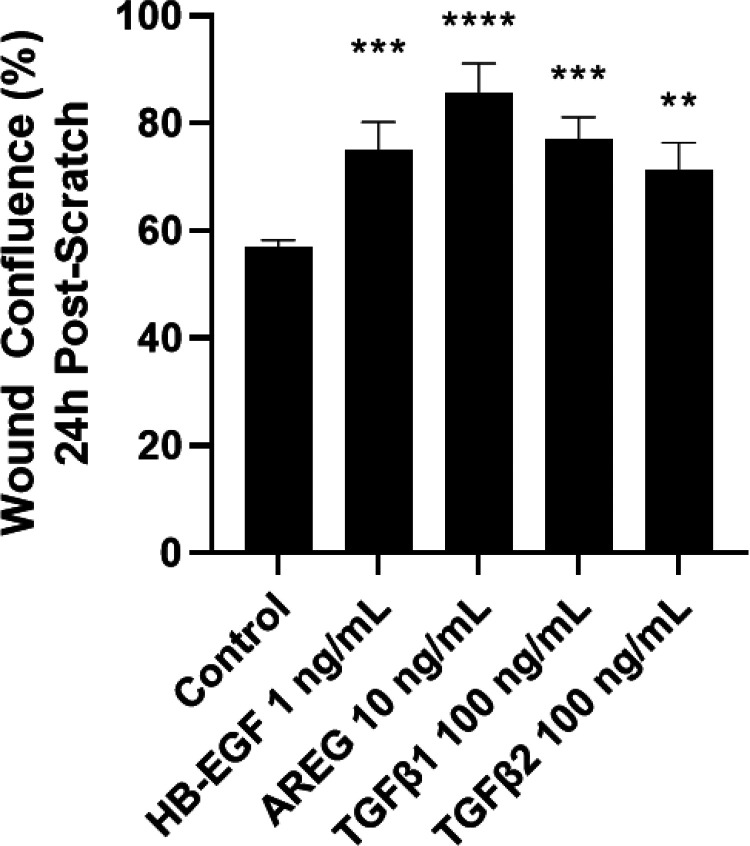
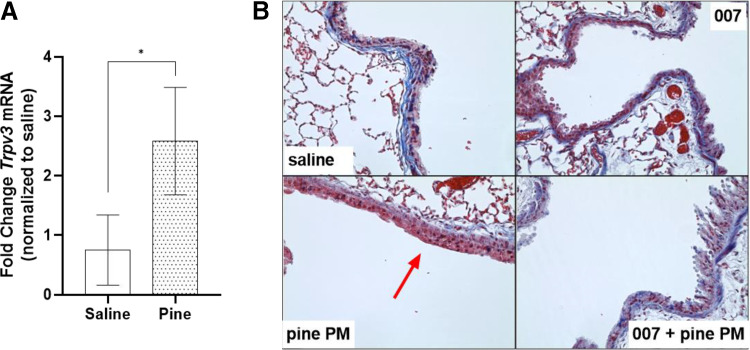
Prior studies revealed increased expression of the transient receptor potential vanilloid-3 (TRPV3) ion channel after wood smoke particulate matter (WSPM) treatment of human bronchial epithelial cells (HBECs). TRPV3 attenuated pathologic endoplasmic reticulum stress and cytotoxicity mediated by transient receptor potential ankyrin-1. Here, the basis for how TRPV3 expression is regulated by cell injury and the effects this has on HBEC physiology and WSPM-induced airway remodeling in mice was investigated. TRPV3 mRNA was rapidly increased in HBECs treated with WSPM and after monolayer damage caused by tryptic disruption, scratch wounding, and cell passaging. TRPV3 mRNA abundance varied with time, and stimulated expression occurred independent of new protein synthesis. Overexpression of TRPV3 in HBECs reduced cell migration and wound repair while enhancing cell adhesion. This phenotype correlated with disrupted mRNA expression of ligands of the epidermal growth factor, tumor growth factor-β, and frizzled receptors. Accordingly, delayed wound repair by TRPV3 overexpressing cells was reversed by growth factor supplementation. In normal HBECs, TRPV3 upregulation was triggered by exogenous growth factor supplementation and was attenuated by inhibitors of growth factor receptor signaling. In mice, subacute oropharyngeal instillation with WSPM also promoted TRPV3 mRNA expression and epithelial remodeling, which was attenuated by TRPV3 antagonist pre- and cotreatment. This latter effect may be the consequence of antagonist-induced TRPV3 expression. These findings provide insights into the roles of TRPV3 in lung epithelial cells under basal and dynamic states, as well as highlight potential roles for TRPV3 ligands in modulating epithelial damage/repair. SIGNIFICANCE STATEMENT: Coordinated epithelial repair is essential for the maintenance of the airways, with deficiencies and exaggerated repair associated with adverse consequences to respiratory health. This study shows that TRPV3, an ion channel, is involved in coordinating repair through integrated repair signaling pathways, wherein TRPV3 expression is upregulated immediately after injury and returns to basal levels as cells complete the repair process. TRPV3 may be a novel target for understanding and/or treating conditions in which airway/lung epithelial repair is not properly orchestrated.
Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics.
Figures







References
-
- Aijima RWang BTakao TMihara HKashio MOhsaki YZhang J-QMizuno ASuzuki MYamashita Y, et al. (2015) The thermosensitive TRPV3 channel contributes to rapid wound healing in oral epithelia. FASEB J 29:182–192 Federation of American Societies for Experimental Biology. - PubMed
-
- Barrow RE, Wang C-Z, Cox RA, Evans MJ (1992) Cellular sequence of tracheal repair in sheep after smoke inhalation injury. Lung 170:331–338. - PubMed
-
- Borbíró ILisztes ETóth BICzifra GOláh ASzöllosi AGSzentandrássy NNánási PPPéter ZPaus R, et al. (2011) Activation of transient receptor potential vanilloid-3 inhibits human hair growth. J Invest Dermatol 131:1605–1614. - PubMed
-
- Bryant JA, Finn RS, Slamon DJ, Cloughesy TF, Charles AC (2004) EGF activates intracellular and intercellular calcium signaling by distinct pathways in tumor cells. Cancer Biol Ther 3:1243–1249 Taylor & Francis. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
