

Codependency and mutual exclusivity for gene community detection from sparse single-cell transcriptome data
- PMID: 34291282
- PMCID: PMC8501962
- DOI: 10.1093/nar/gkab601
Codependency and mutual exclusivity for gene community detection from sparse single-cell transcriptome data
Abstract
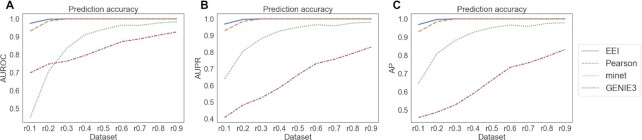
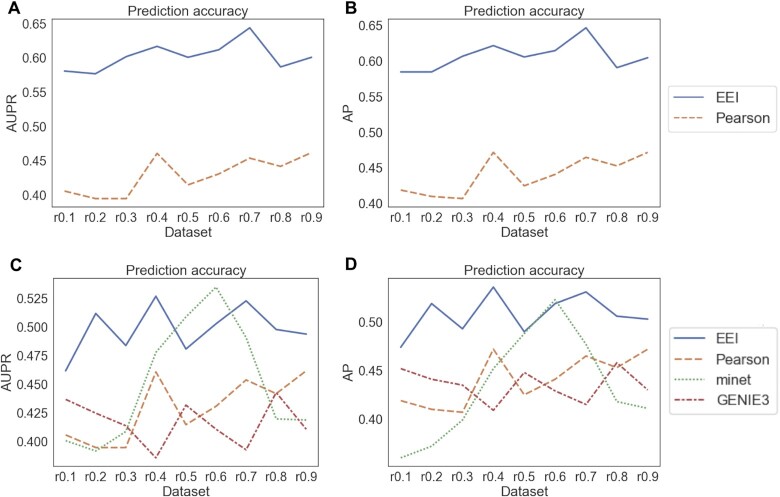
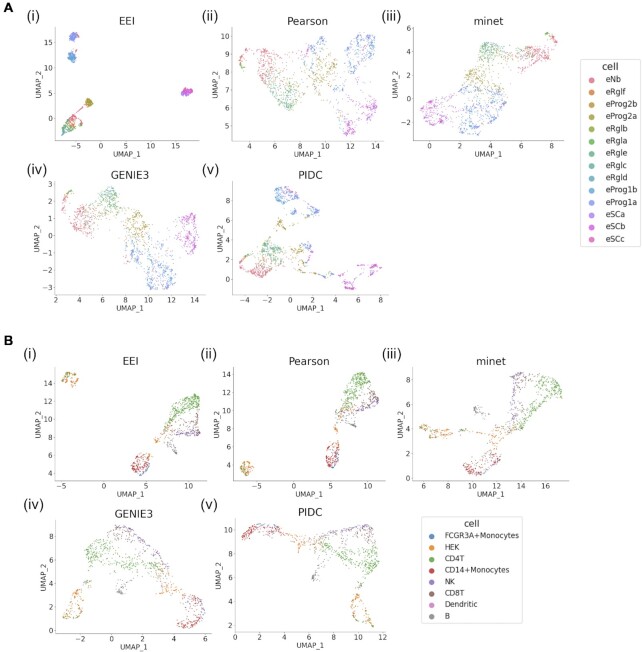
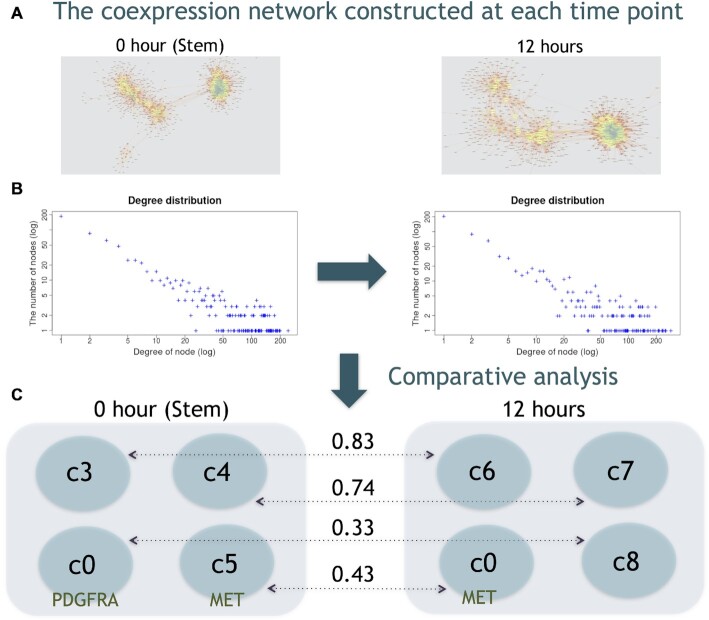
Single-cell RNA-seq (scRNA-seq) can be used to characterize cellular heterogeneity in thousands of cells. The reconstruction of a gene network based on coexpression patterns is a fundamental task in scRNA-seq analyses, and the mutual exclusivity of gene expression can be critical for understanding such heterogeneity. Here, we propose an approach for detecting communities from a genetic network constructed on the basis of coexpression properties. The community-based comparison of multiple coexpression networks enables the identification of functionally related gene clusters that cannot be fully captured through differential gene expression-based analysis. We also developed a novel metric referred to as the exclusively expressed index (EEI) that identifies mutually exclusive gene pairs from sparse scRNA-seq data. EEI quantifies and ranks the exclusive expression levels of all gene pairs from binary expression patterns while maintaining robustness against a low sequencing depth. We applied our methods to glioblastoma scRNA-seq data and found that gene communities were partially conserved after serum stimulation despite a considerable number of differentially expressed genes. We also demonstrate that the identification of mutually exclusive gene sets with EEI can improve the sensitivity of capturing cellular heterogeneity. Our methods complement existing approaches and provide new biological insights, even for a large, sparse dataset, in the single-cell analysis field.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

Similar articles
-
scNPF: an integrative framework assisted by network propagation and network fusion for preprocessing of single-cell RNA-seq data.BMC Genomics. 2019 May 8;20(1):347. doi: 10.1186/s12864-019-5747-5. BMC Genomics. 2019. PMID: 31068142 Free PMC article.
-
Detection of high variability in gene expression from single-cell RNA-seq profiling.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):508. doi: 10.1186/s12864-016-2897-6. BMC Genomics. 2016. PMID: 27556924 Free PMC article.
-
Cell Heterogeneity Analysis in Single-Cell RNA-seq Data Using Mixture Exponential Graph and Markov Random Field Model.Biomed Res Int. 2021 May 22;2021:9919080. doi: 10.1155/2021/9919080. eCollection 2021. Biomed Res Int. 2021. PMID: 34095314 Free PMC article.
-
Single-Cell Sequencing Methodologies: From Transcriptome to Multi-Dimensional Measurement.Small Methods. 2021 Jun;5(6):e2100111. doi: 10.1002/smtd.202100111. Epub 2021 Apr 17. Small Methods. 2021. PMID: 34927917 Review.
-
Single-cell RNA sequencing to study vascular diversity and function.Curr Opin Hematol. 2021 May 1;28(3):221-229. doi: 10.1097/MOH.0000000000000651. Curr Opin Hematol. 2021. PMID: 33714967 Free PMC article. Review.
Cited by
-
Glioblastoma Stem Cells-Useful Tools in the Battle against Cancer.Int J Mol Sci. 2022 Apr 21;23(9):4602. doi: 10.3390/ijms23094602. Int J Mol Sci. 2022. PMID: 35562993 Free PMC article. Review.
-
Multi-level cellular and functional annotation of single-cell transcriptomes using scPipeline.Commun Biol. 2022 Oct 28;5(1):1142. doi: 10.1038/s42003-022-04093-2. Commun Biol. 2022. PMID: 36307536 Free PMC article.
-
Using Sequence Analyses to Quantitatively Measure Oropharyngeal Swallowing Temporality in Point-of-Care Ultrasound Examinations: A Pilot Study.J Clin Med. 2024 Apr 15;13(8):2288. doi: 10.3390/jcm13082288. J Clin Med. 2024. PMID: 38673561 Free PMC article.
-
Gene2role: a role-based gene embedding method for comparative analysis of signed gene regulatory networks.BMC Bioinformatics. 2025 May 24;26(1):134. doi: 10.1186/s12859-025-06128-x. BMC Bioinformatics. 2025. PMID: 40413377 Free PMC article.
-
Integrated single-cell transcriptomic and epigenetic study of cell state transition and lineage commitment in embryonic mouse cerebellum.Sci Adv. 2022 Apr;8(13):eabl9156. doi: 10.1126/sciadv.abl9156. Epub 2022 Apr 1. Sci Adv. 2022. PMID: 35363520 Free PMC article.
References
-
- Akutsu T., Miyano S., Kuhara S.. Inferring qualitative relations in genetic networks and metabolic pathways. Bioinformatics. 2000; 16:727–734. - PubMed
-
- Hickman G.J., Hodgman T.C.. Inference of gene regulatory networks using boolean-network inference methods. J. Bioinform. Comput. B. 2009; 7:1013–1029. - PubMed
-
- Barman S., Kwon Y.K.. A Boolean network inference from time-series gene expression data using a genetic algorithm. Bioinformatics. 2018; 34:1927–1933. - PubMed
-
- Penfold C.A., Shifaz A., Brown P.E., Nicholson A., Wild D.L.. CSI: a nonparametric Bayesian approach to network inference from multiple perturbed time series gene expression data. Stat. Appl. Genet. Mol. Biol. 2015; 14:307–310. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
