

Ongoing global and regional adaptive evolution of SARS-CoV-2
- PMID: 34292871
- PMCID: PMC8307621
- DOI: 10.1073/pnas.2104241118
Ongoing global and regional adaptive evolution of SARS-CoV-2
Abstract
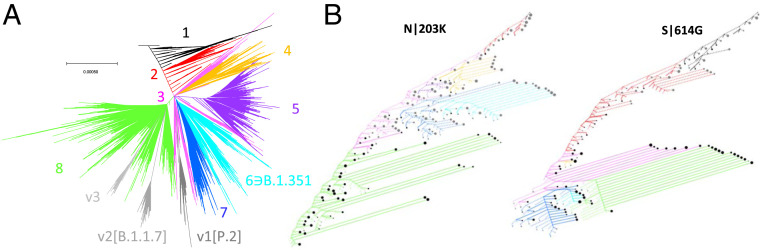
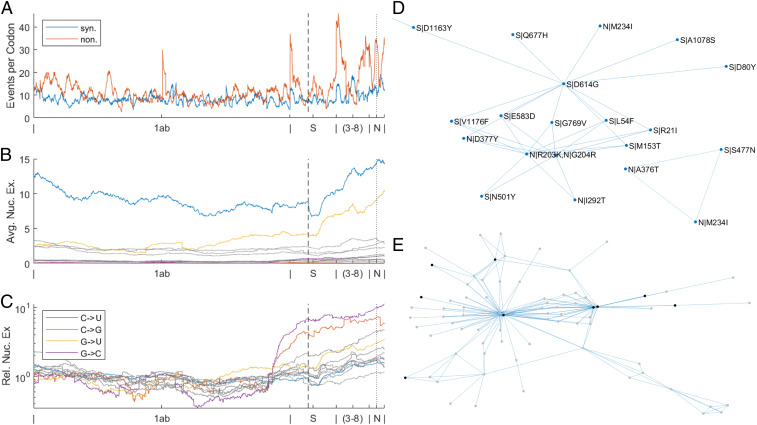
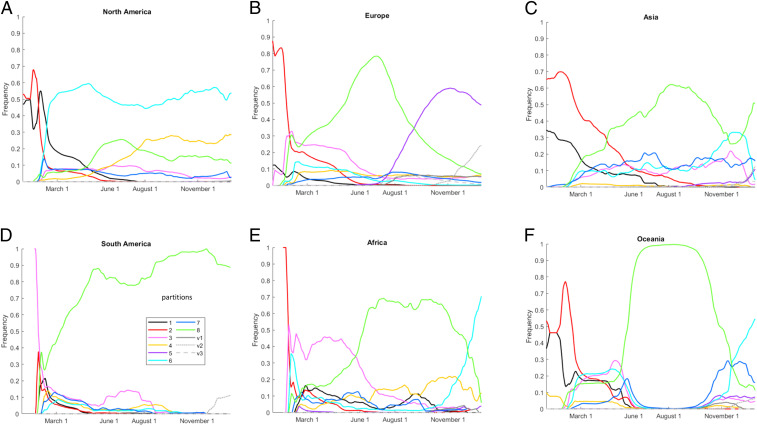
Understanding the trends in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) evolution is paramount to control the COVID-19 pandemic. We analyzed more than 300,000 high-quality genome sequences of SARS-CoV-2 variants available as of January 2021. The results show that the ongoing evolution of SARS-CoV-2 during the pandemic is characterized primarily by purifying selection, but a small set of sites appear to evolve under positive selection. The receptor-binding domain of the spike protein and the region of the nucleocapsid protein associated with nuclear localization signals (NLS) are enriched with positively selected amino acid replacements. These replacements form a strongly connected network of apparent epistatic interactions and are signatures of major partitions in the SARS-CoV-2 phylogeny. Virus diversity within each geographic region has been steadily growing for the entirety of the pandemic, but analysis of the phylogenetic distances between pairs of regions reveals four distinct periods based on global partitioning of the tree and the emergence of key mutations. The initial period of rapid diversification into region-specific phylogenies that ended in February 2020 was followed by a major extinction event and global homogenization concomitant with the spread of D614G in the spike protein, ending in March 2020. The NLS-associated variants across multiple partitions rose to global prominence in March to July, during a period of stasis in terms of interregional diversity. Finally, beginning in July 2020, multiple mutations, some of which have since been demonstrated to enable antibody evasion, began to emerge associated with ongoing regional diversification, which might be indicative of speciation.
Keywords: SARS-Cov-2; ancestral reconstruction; epistasis; globalization; phylogeny.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures


Update of
-
Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2.bioRxiv [Preprint]. 2021 Mar 2:2020.10.12.336644. doi: 10.1101/2020.10.12.336644. bioRxiv. 2021. Update in: Proc Natl Acad Sci U S A. 2021 Jul 20;118(29):e2104241118. doi: 10.1073/pnas.2104241118. PMID: 33083804 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
