

Highly accurate protein structure prediction for the human proteome
- PMID: 34293799
- PMCID: PMC8387240
- DOI: 10.1038/s41586-021-03828-1
Highly accurate protein structure prediction for the human proteome
Abstract
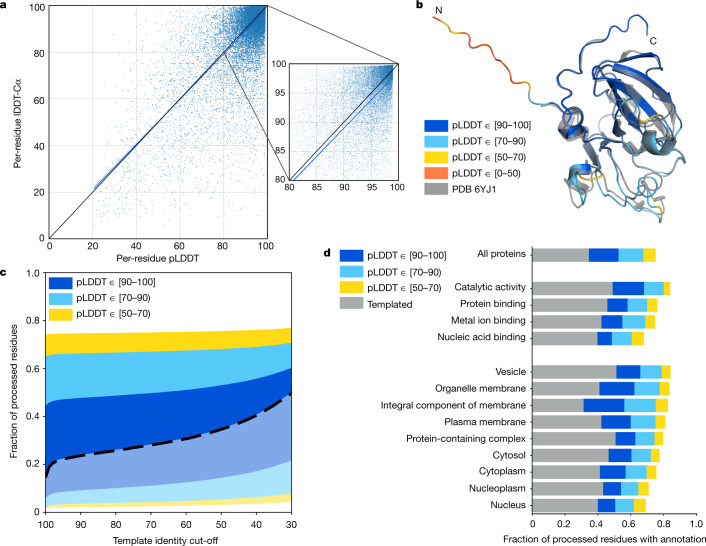
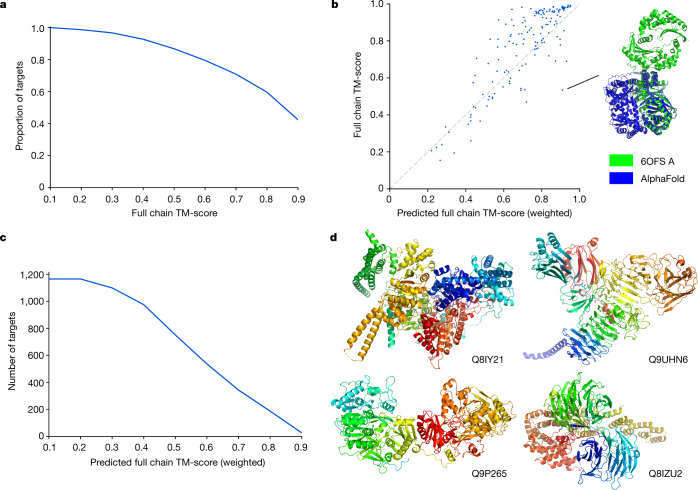
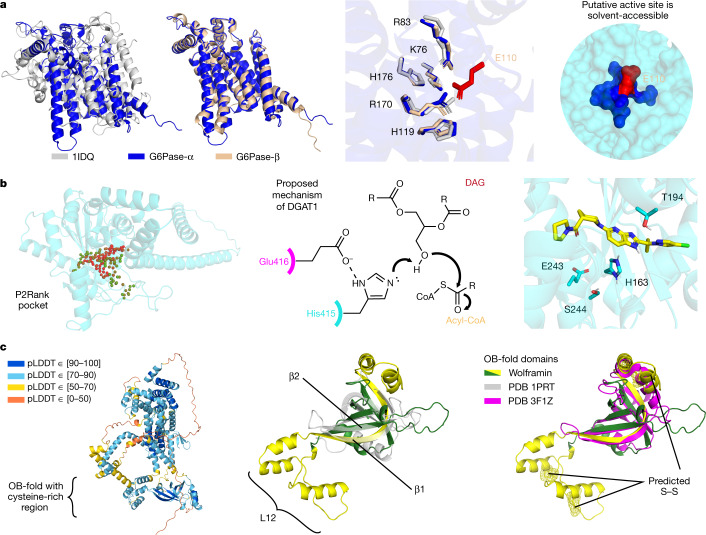
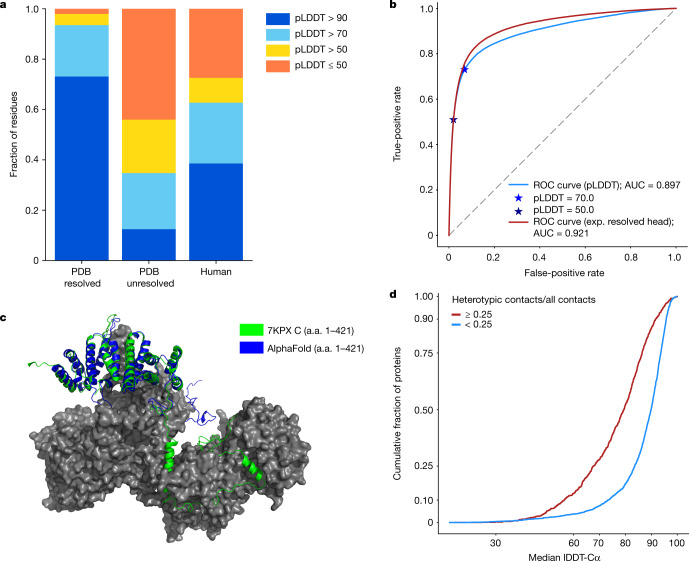
Protein structures can provide invaluable information, both for reasoning about biological processes and for enabling interventions such as structure-based drug development or targeted mutagenesis. After decades of effort, 17% of the total residues in human protein sequences are covered by an experimentally determined structure1. Here we markedly expand the structural coverage of the proteome by applying the state-of-the-art machine learning method, AlphaFold2, at a scale that covers almost the entire human proteome (98.5% of human proteins). The resulting dataset covers 58% of residues with a confident prediction, of which a subset (36% of all residues) have very high confidence. We introduce several metrics developed by building on the AlphaFold model and use them to interpret the dataset, identifying strong multi-domain predictions as well as regions that are likely to be disordered. Finally, we provide some case studies to illustrate how high-quality predictions could be used to generate biological hypotheses. We are making our predictions freely available to the community and anticipate that routine large-scale and high-accuracy structure prediction will become an important tool that will allow new questions to be addressed from a structural perspective.
© 2021. The Author(s).
Conflict of interest statement
J.J., R.E., A. Pritzel, T.G., M.F., O.R., R.B., A. Bridgland, S.A.A.K., D.R. and A.W.S. have filed non-provisional patent applications 16/701,070, PCT/EP2020/084238, and provisional patent applications 63/107,362, 63/118,917, 63/118,918, 63/118,921 and 63/118,919, each in the name of DeepMind Technologies Limited, each pending, relating to machine learning for predicting protein structures. E.B. is a paid consultant to Oxford Nanopore and Dovetail Inc, which are genomics companies. The other authors declare no competing interests.
Figures
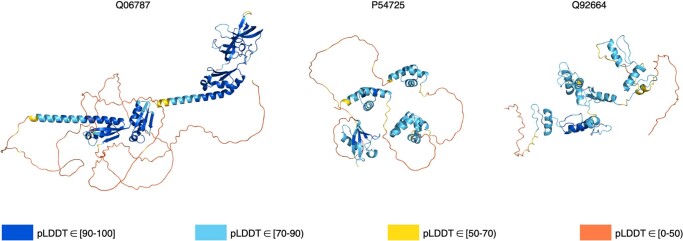
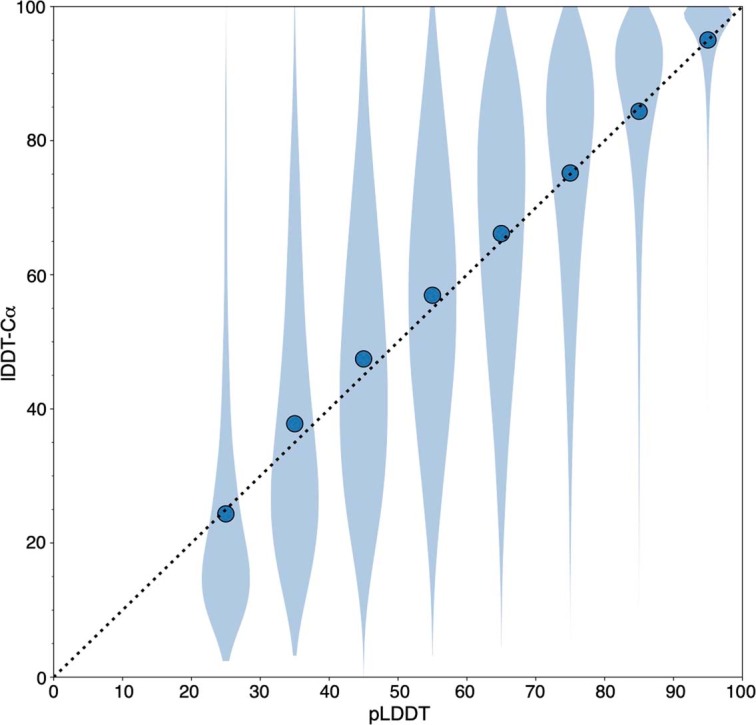
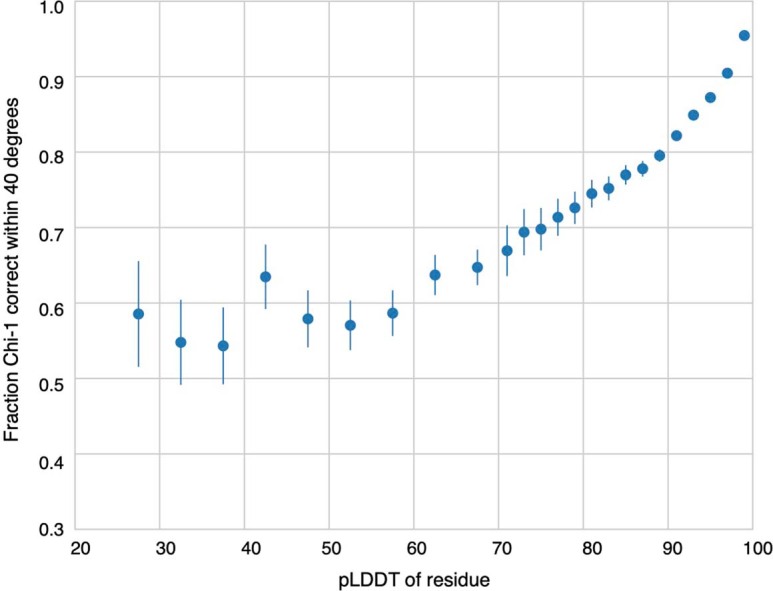
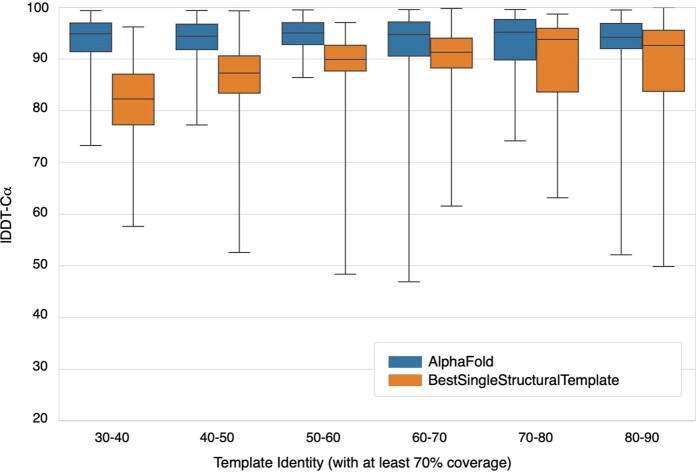
Comment in
-
Protein-structure prediction revolutionized.Nature. 2021 Aug;596(7873):487-488. doi: 10.1038/d41586-021-02265-4. Nature. 2021. PMID: 34426694 No abstract available.
-
AlphaFold developers win US$3-million Breakthrough Prize.Nature. 2022 Sep;609(7929):889. doi: 10.1038/d41586-022-02999-9. Nature. 2022. PMID: 36138210 No abstract available.
References
-
- SWISS-MODEL. Homo sapiens (human). https://swissmodel.expasy.org/repository/species/9606 (2021).
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
