

Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA
- PMID: 34294911
- PMCID: PMC8678141
- DOI: 10.1038/s41587-021-00981-w
Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA
Abstract
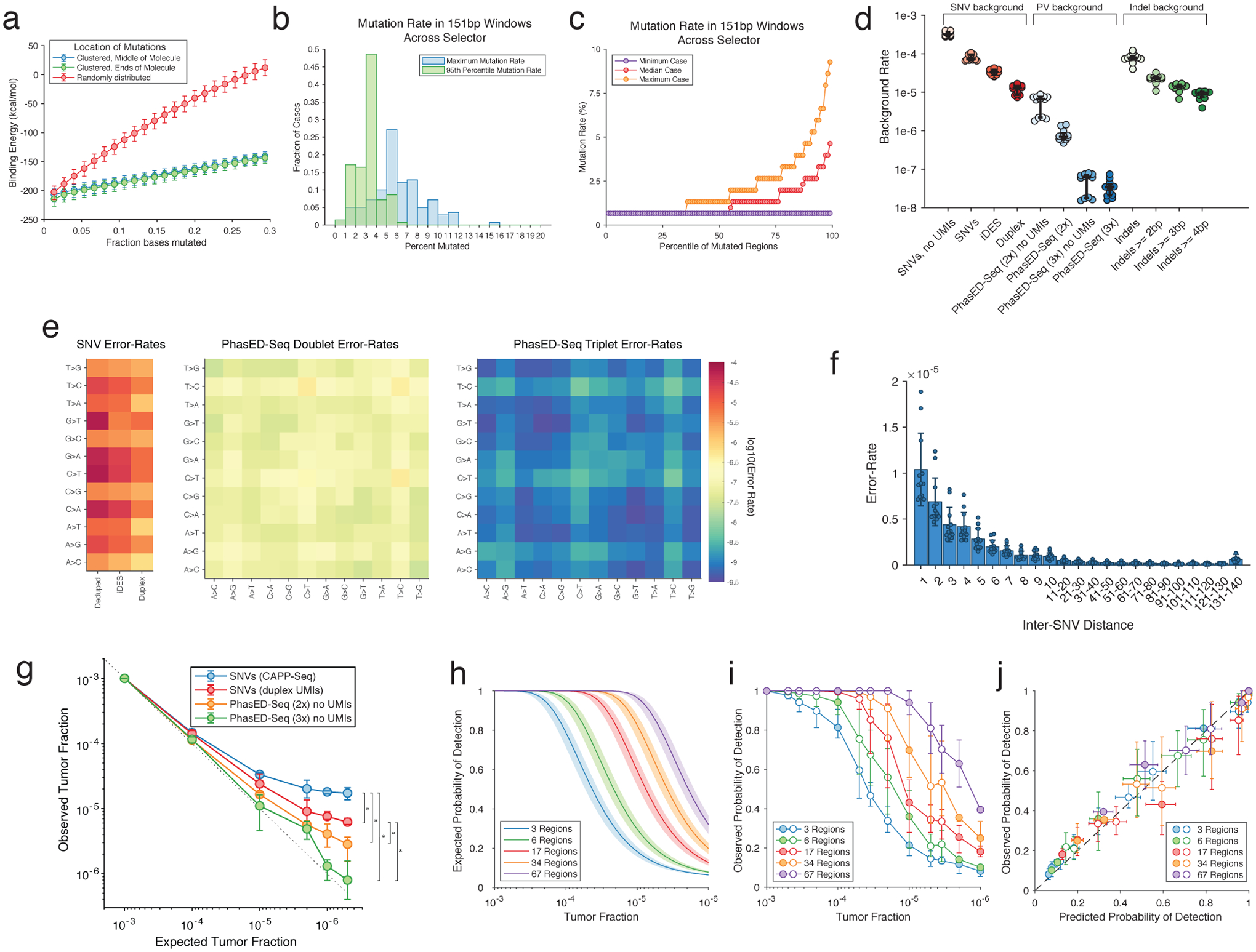
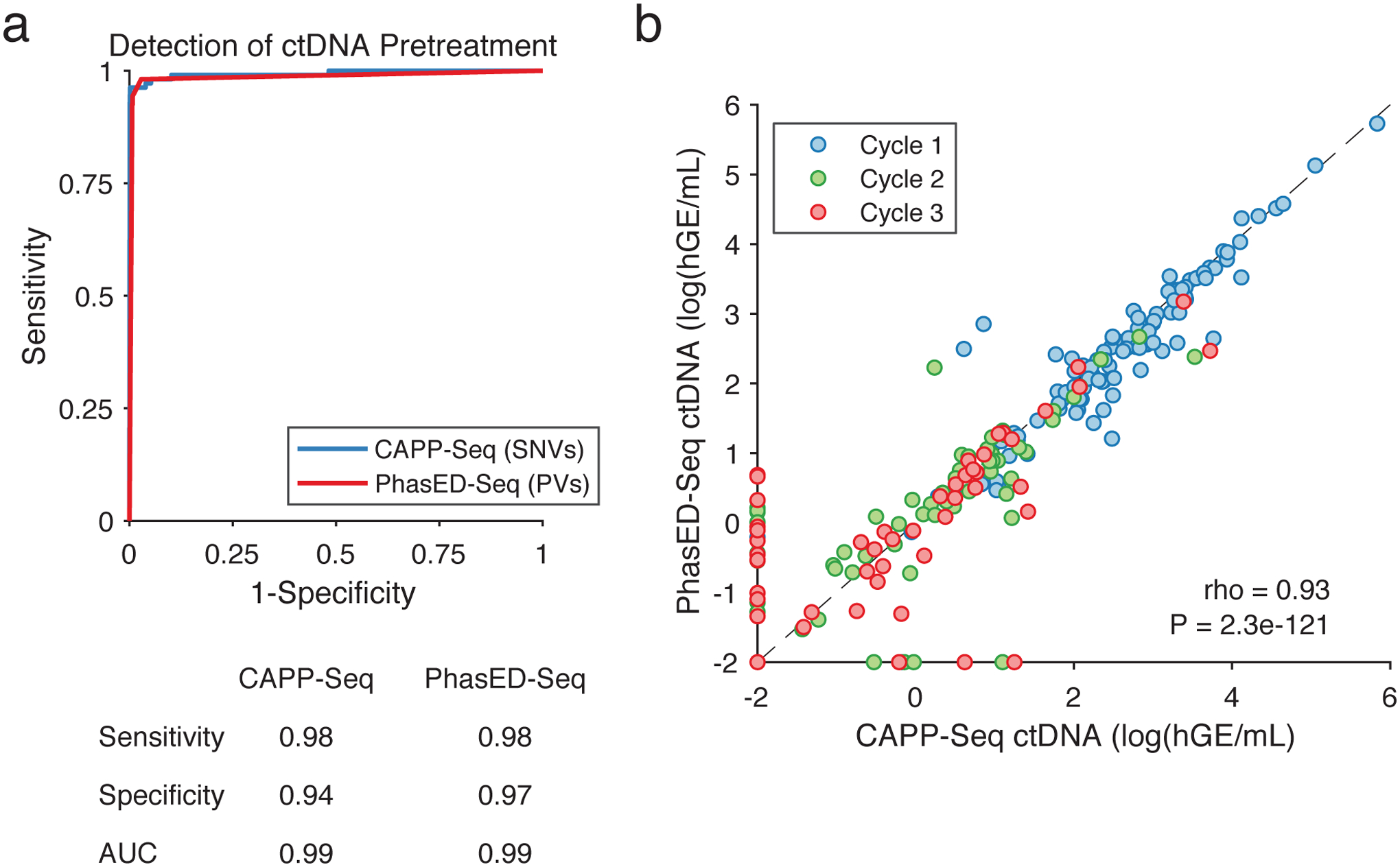
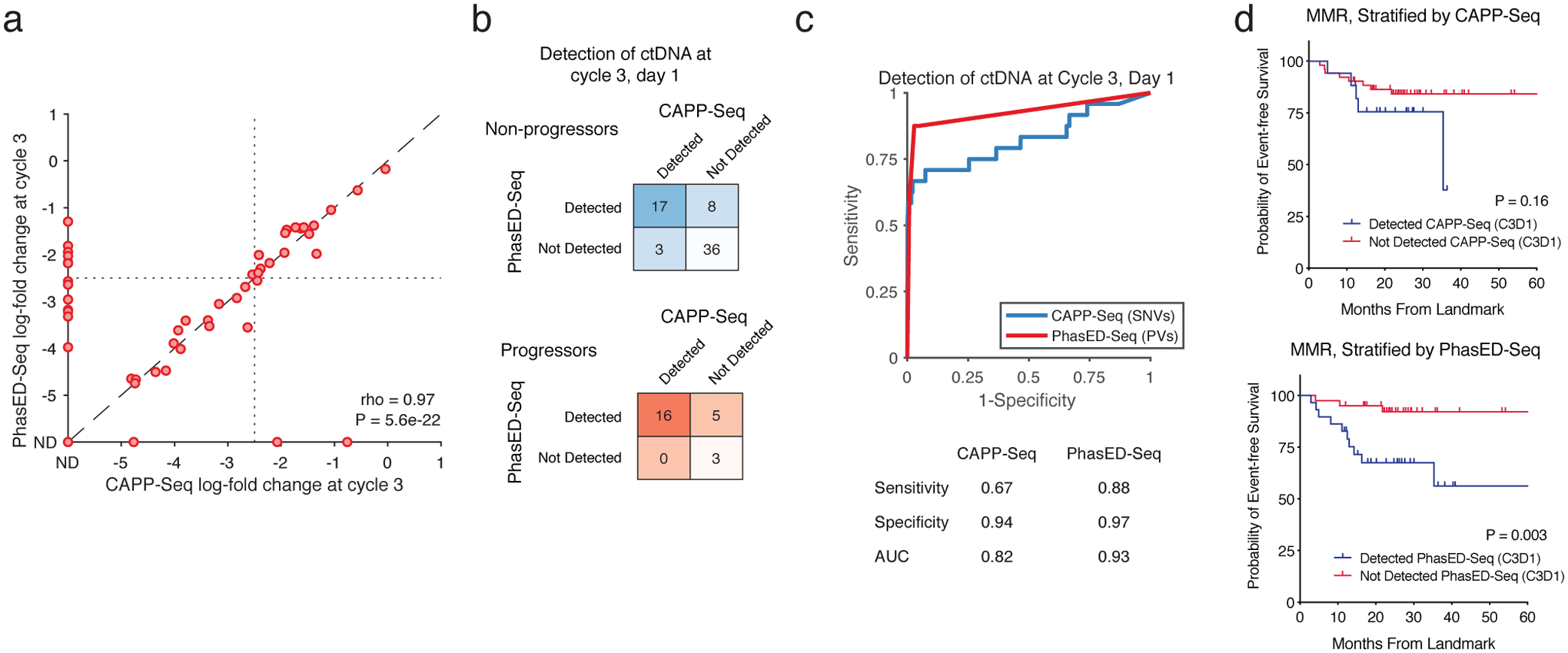
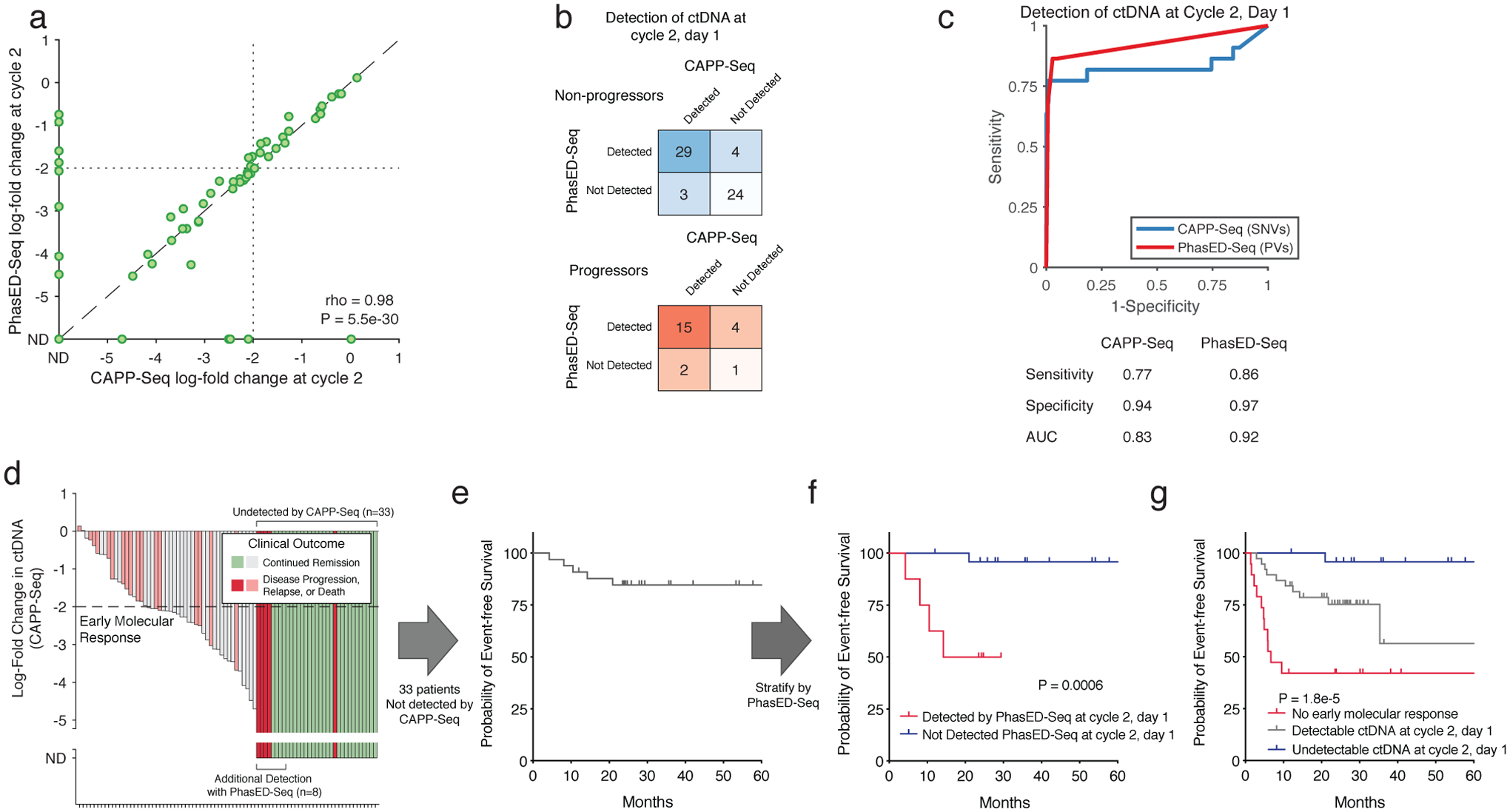
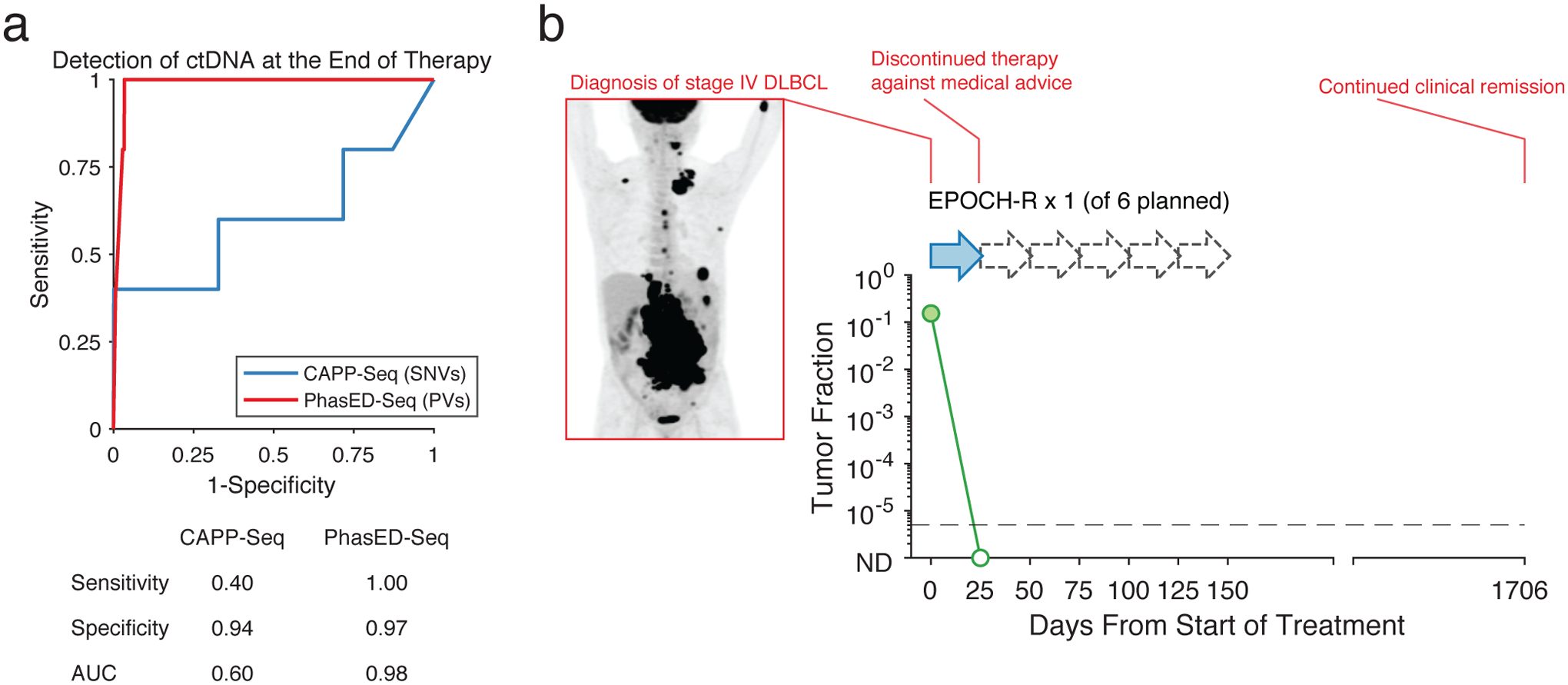
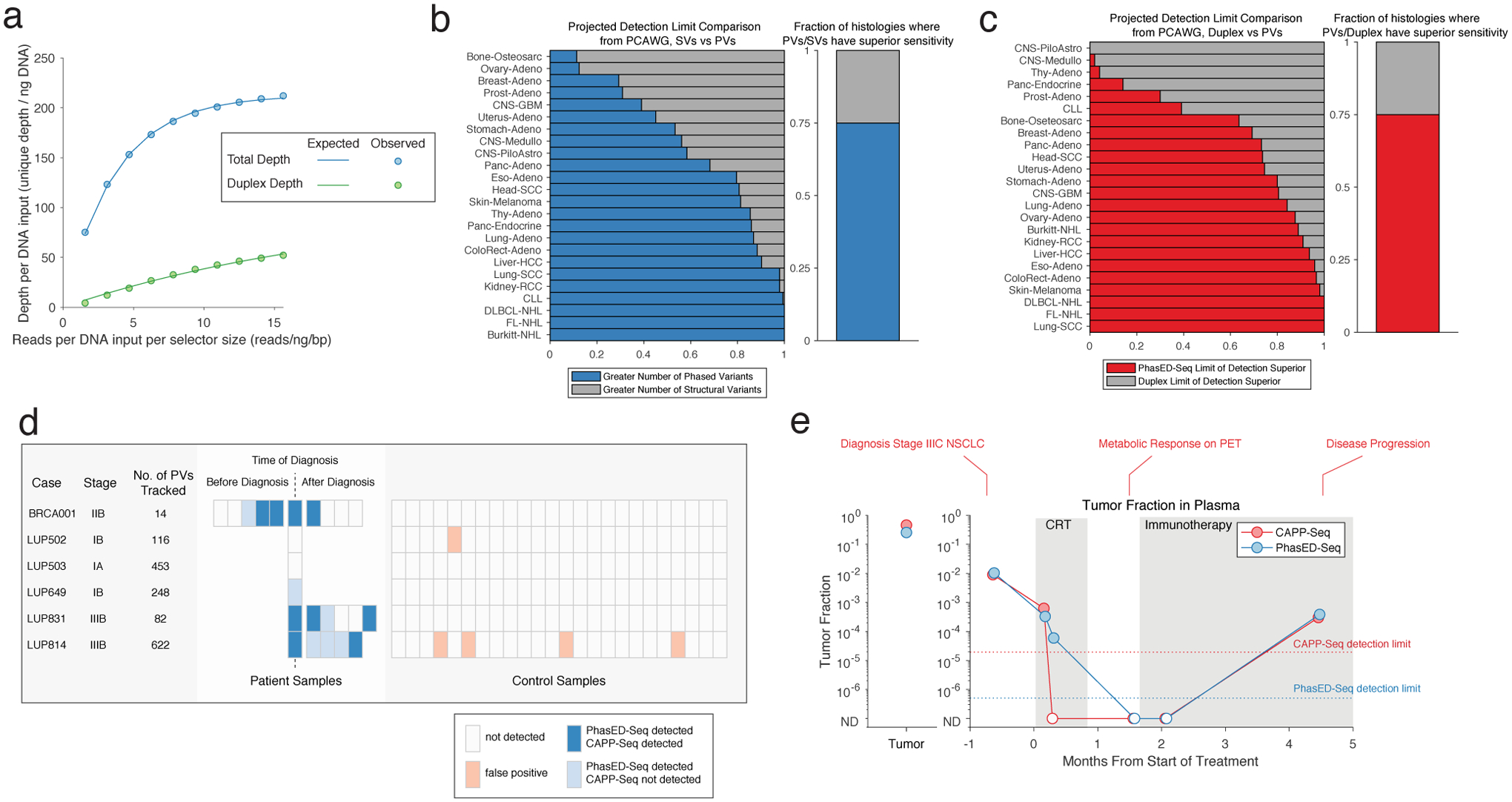
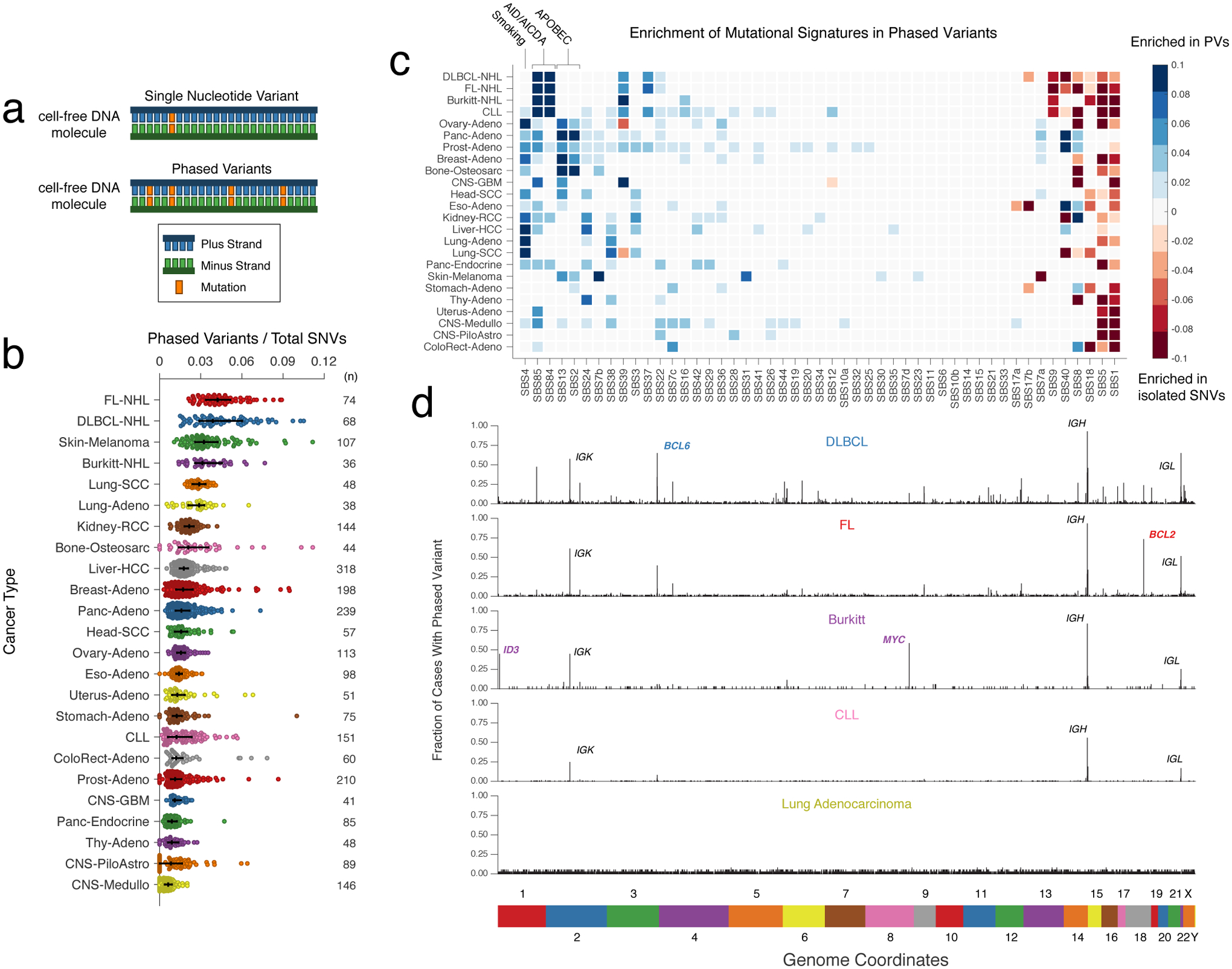
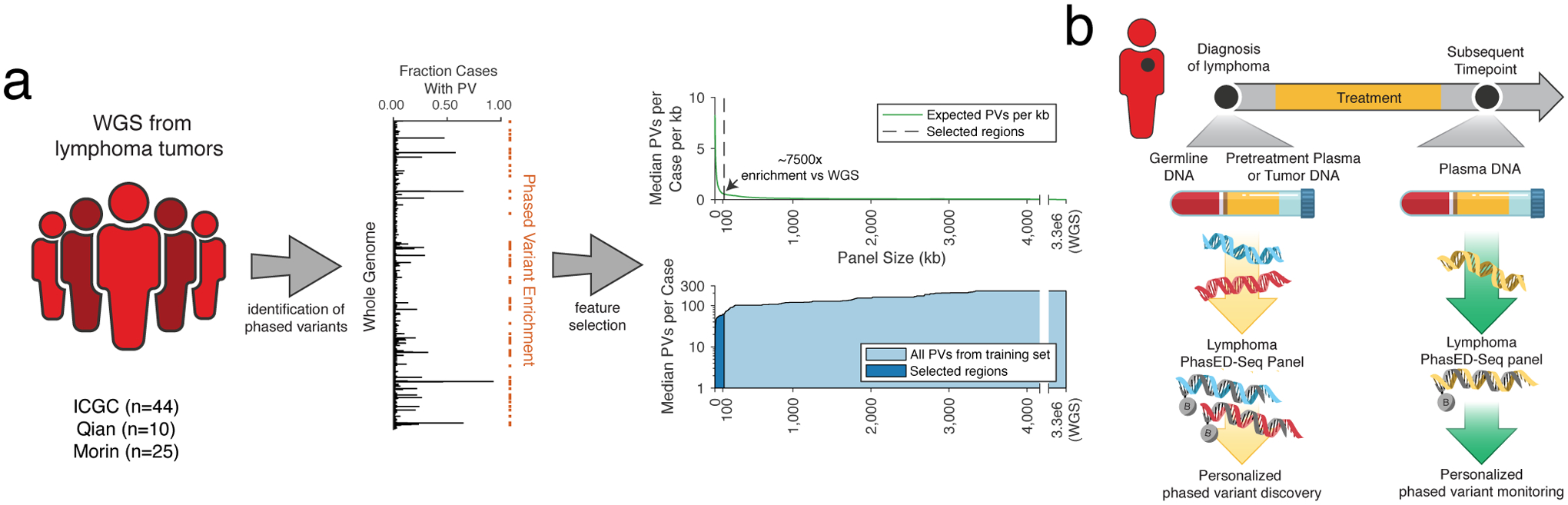
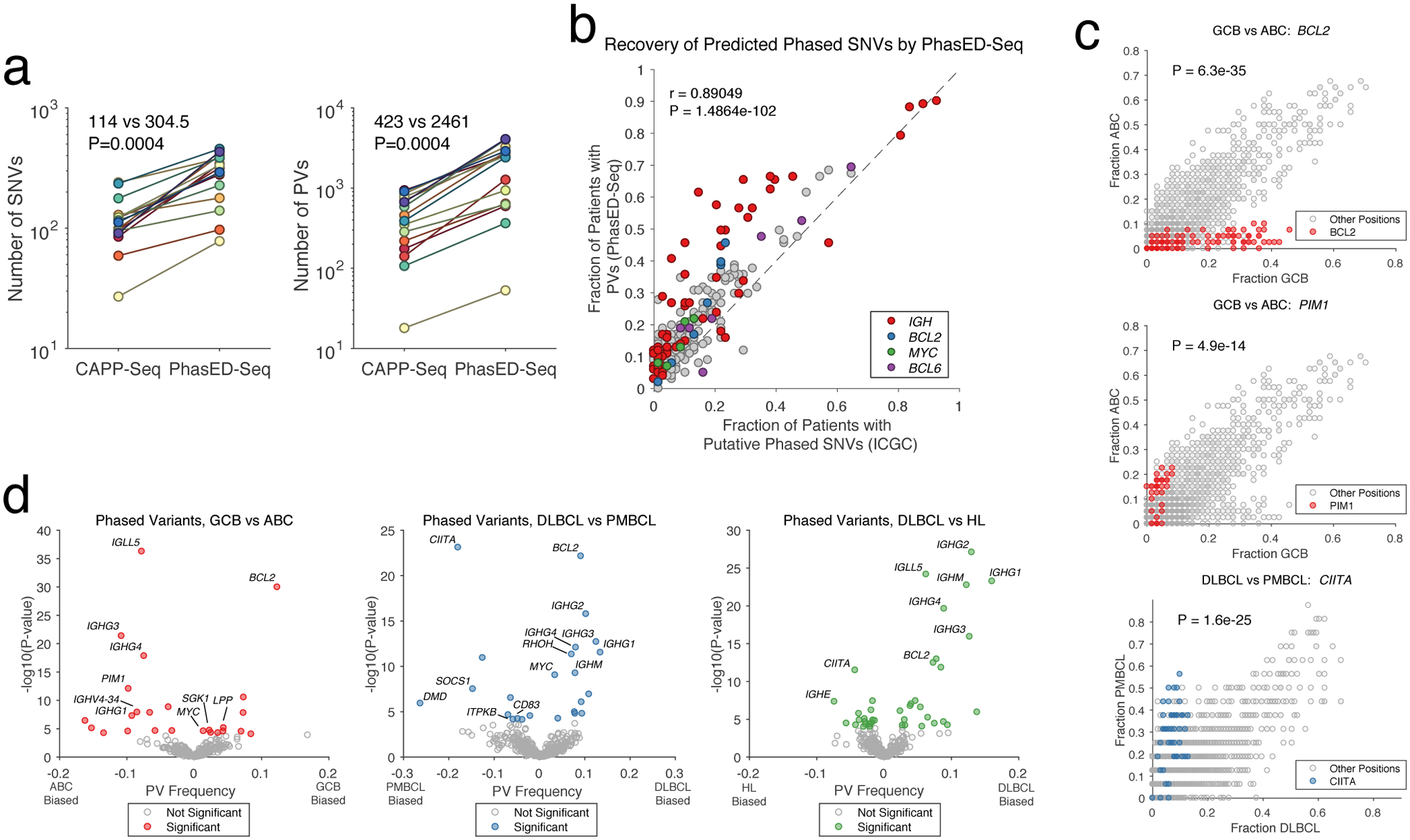
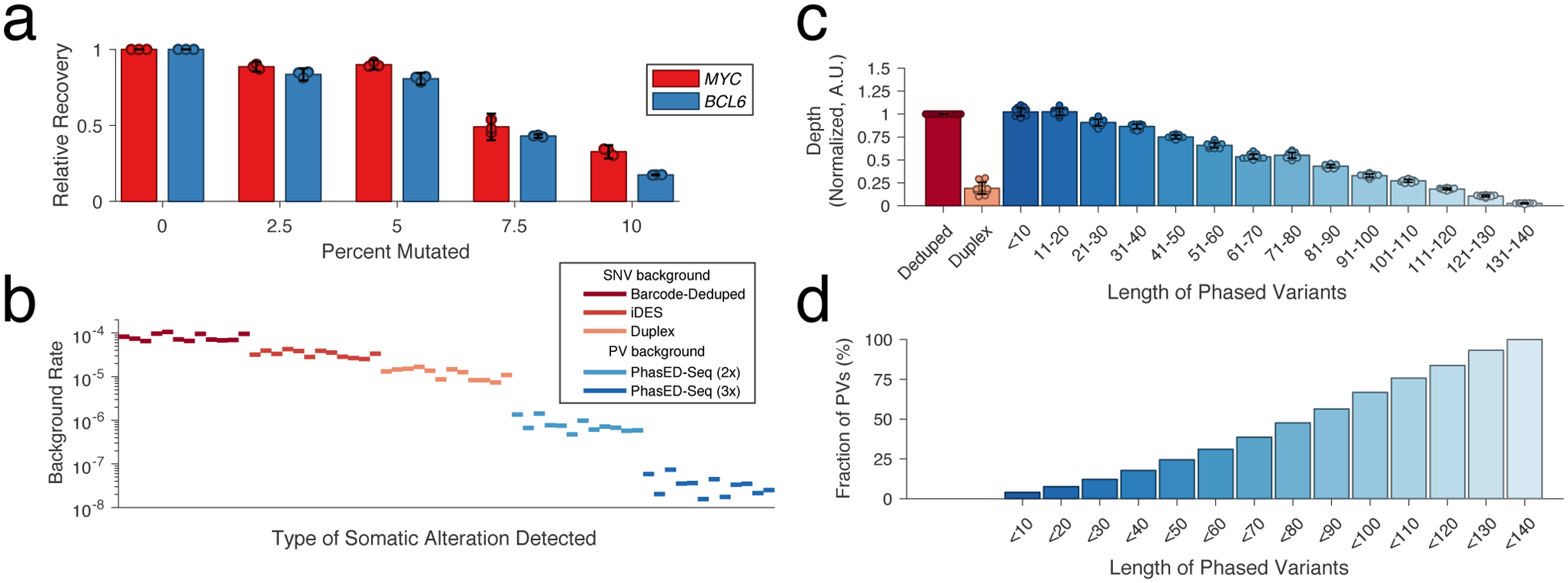
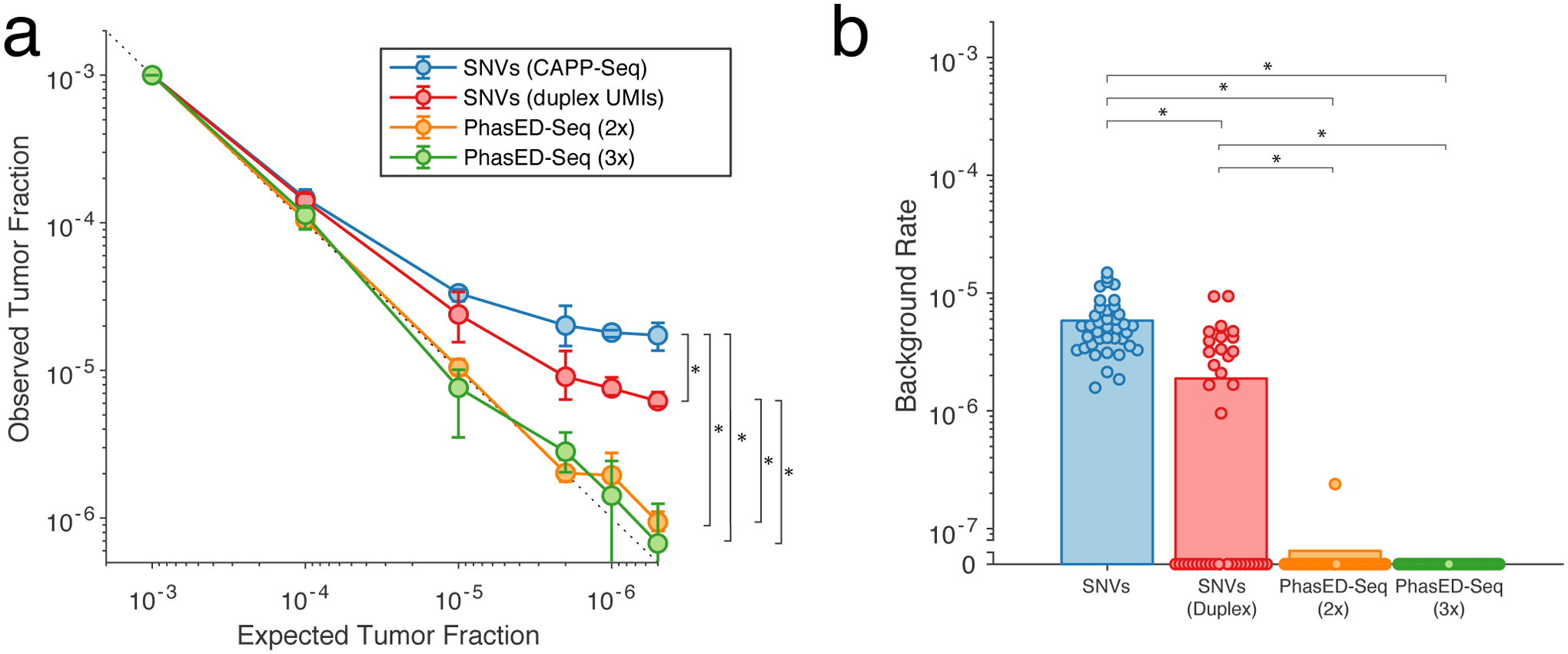
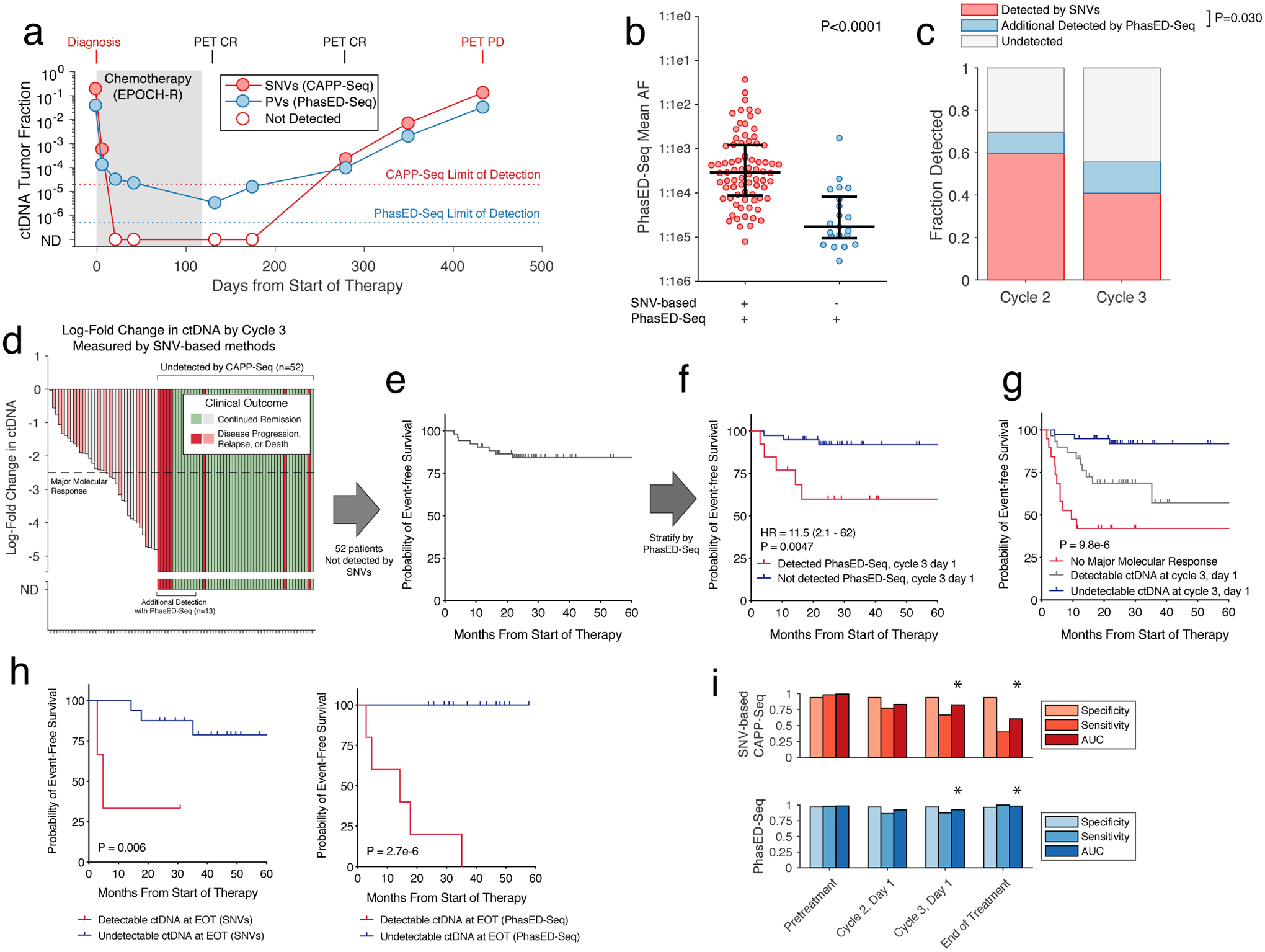
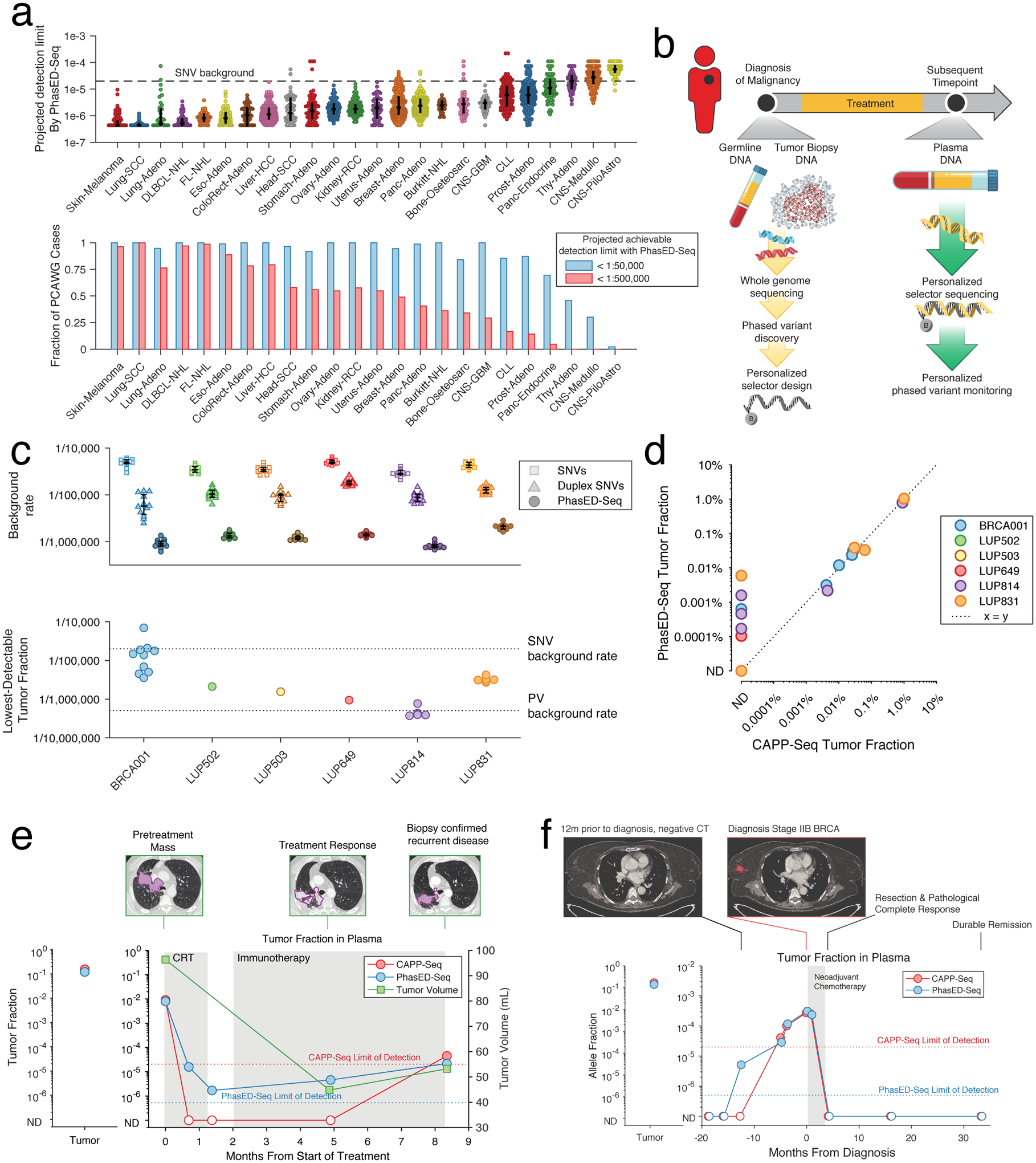
Circulating tumor-derived DNA (ctDNA) is an emerging biomarker for many cancers, but the limited sensitivity of current detection methods reduces its utility for diagnosing minimal residual disease. Here we describe phased variant enrichment and detection sequencing (PhasED-seq), a method that uses multiple somatic mutations in individual DNA fragments to improve the sensitivity of ctDNA detection. Leveraging whole-genome sequences from 2,538 tumors, we identify phased variants and their associations with mutational signatures. We show that even without molecular barcodes, the limits of detection of PhasED-seq outperform prior methods, including duplex barcoding, allowing ctDNA detection in the ppm range in participant samples. We profiled 678 specimens from 213 participants with B cell lymphomas, including serial cell-free DNA samples before and during therapy for diffuse large B cell lymphoma. In participants with undetectable ctDNA after two cycles of therapy using a next-generation sequencing-based approach termed cancer personalized profiling by deep sequencing, an additional 25% have ctDNA detectable by PhasED-seq and have worse outcomes. Finally, we demonstrate the application of PhasED-seq to solid tumors.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
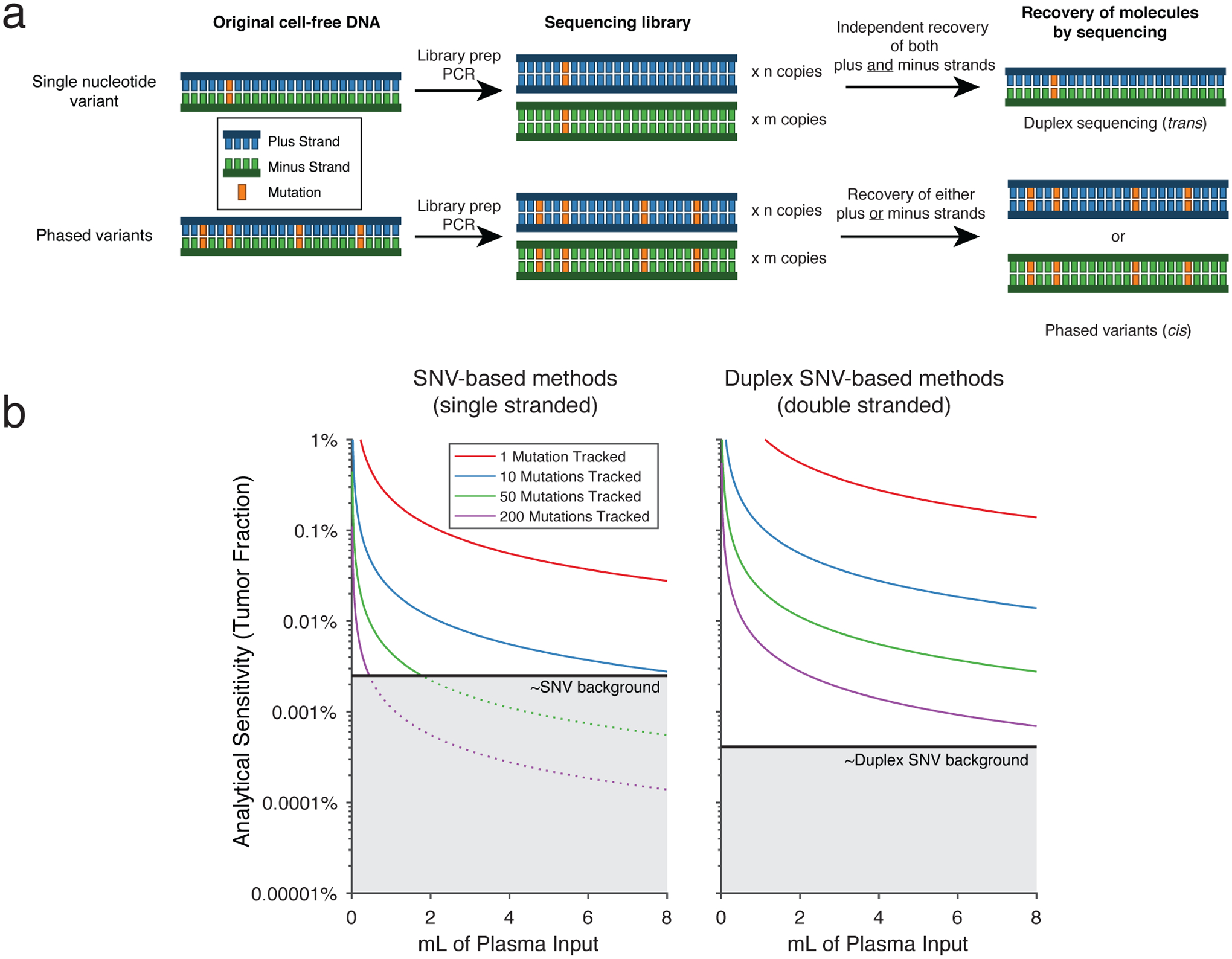
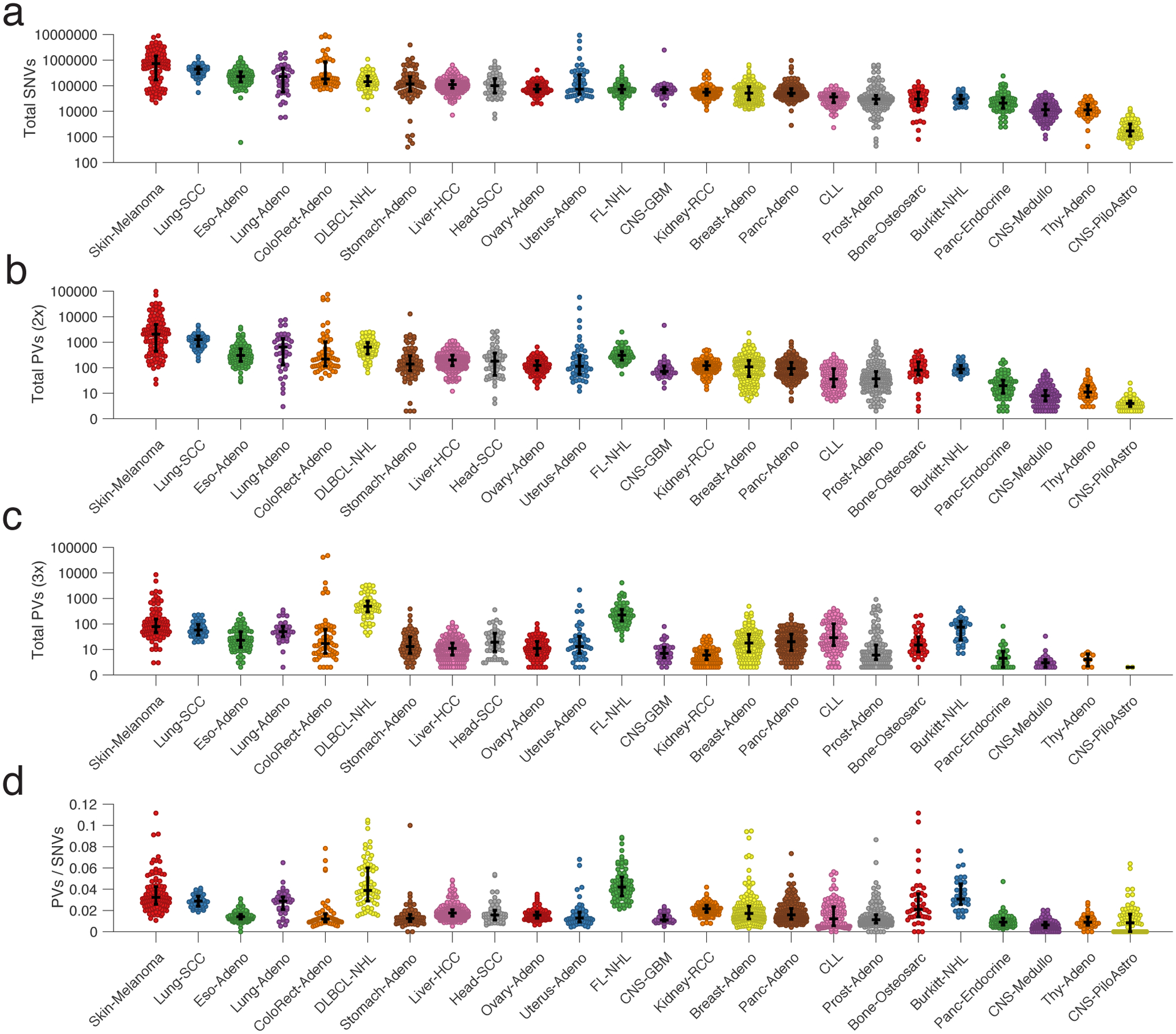
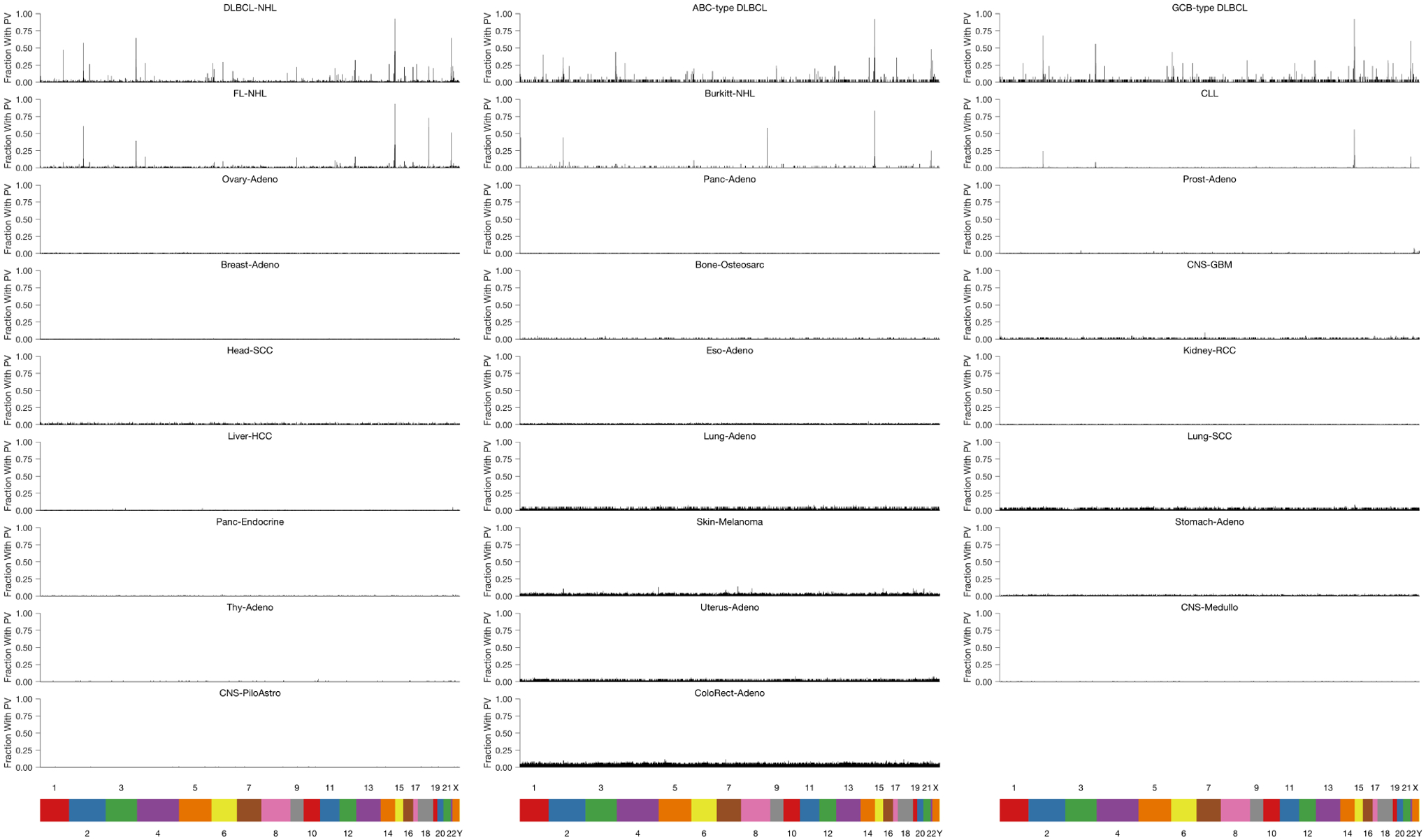
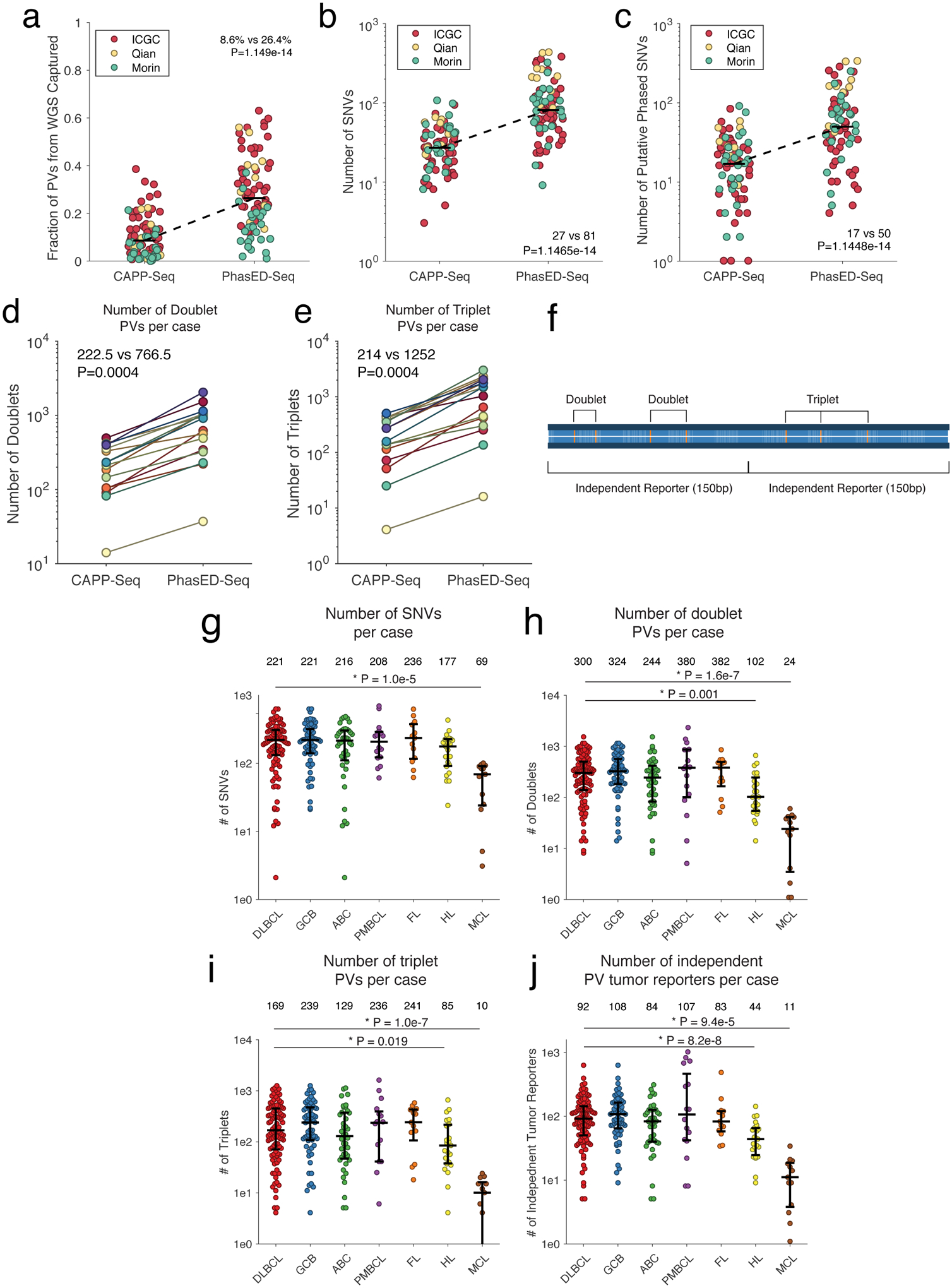
Comment in
-
Lightning does strike twice: leveraging phased variants to enhance minimal residual disease detection.Med. 2021 Oct 8;2(10):1114-1116. doi: 10.1016/j.medj.2021.09.005. Med. 2021. PMID: 35590202
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
