

Baricitinib, a JAK-STAT Inhibitor, Reduces the Cellular Toxicity of the Farnesyltransferase Inhibitor Lonafarnib in Progeria Cells
- PMID: 34299092
- PMCID: PMC8307450
- DOI: 10.3390/ijms22147474
Baricitinib, a JAK-STAT Inhibitor, Reduces the Cellular Toxicity of the Farnesyltransferase Inhibitor Lonafarnib in Progeria Cells
Abstract
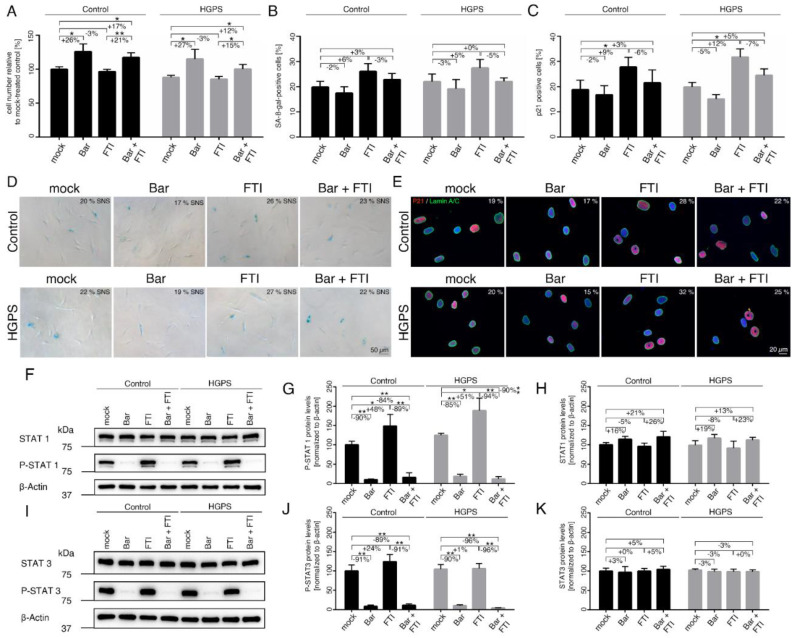
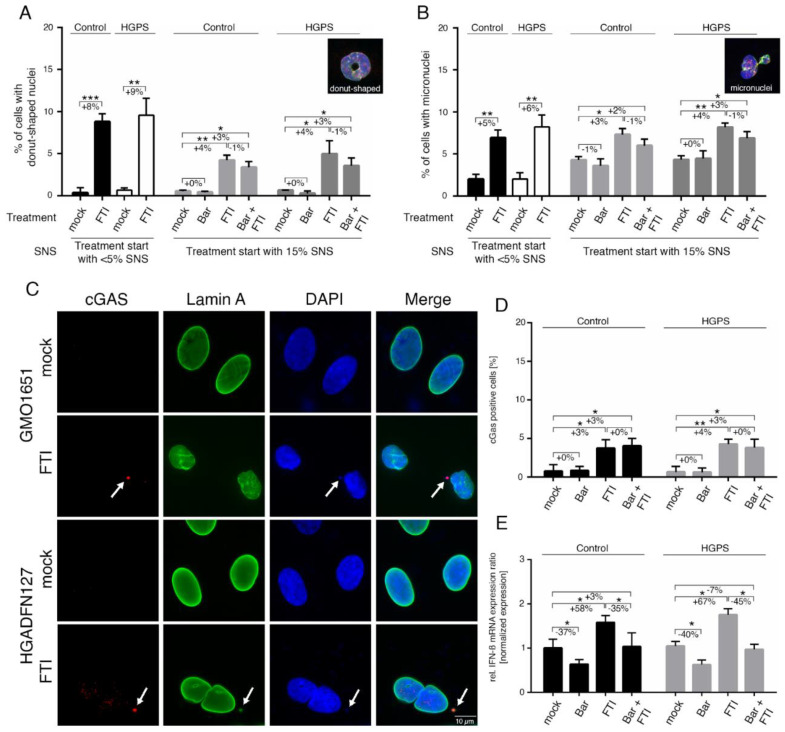
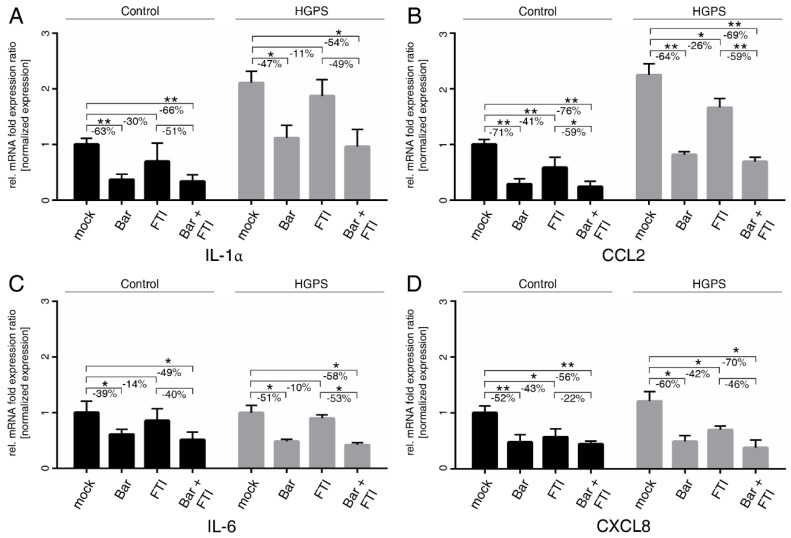
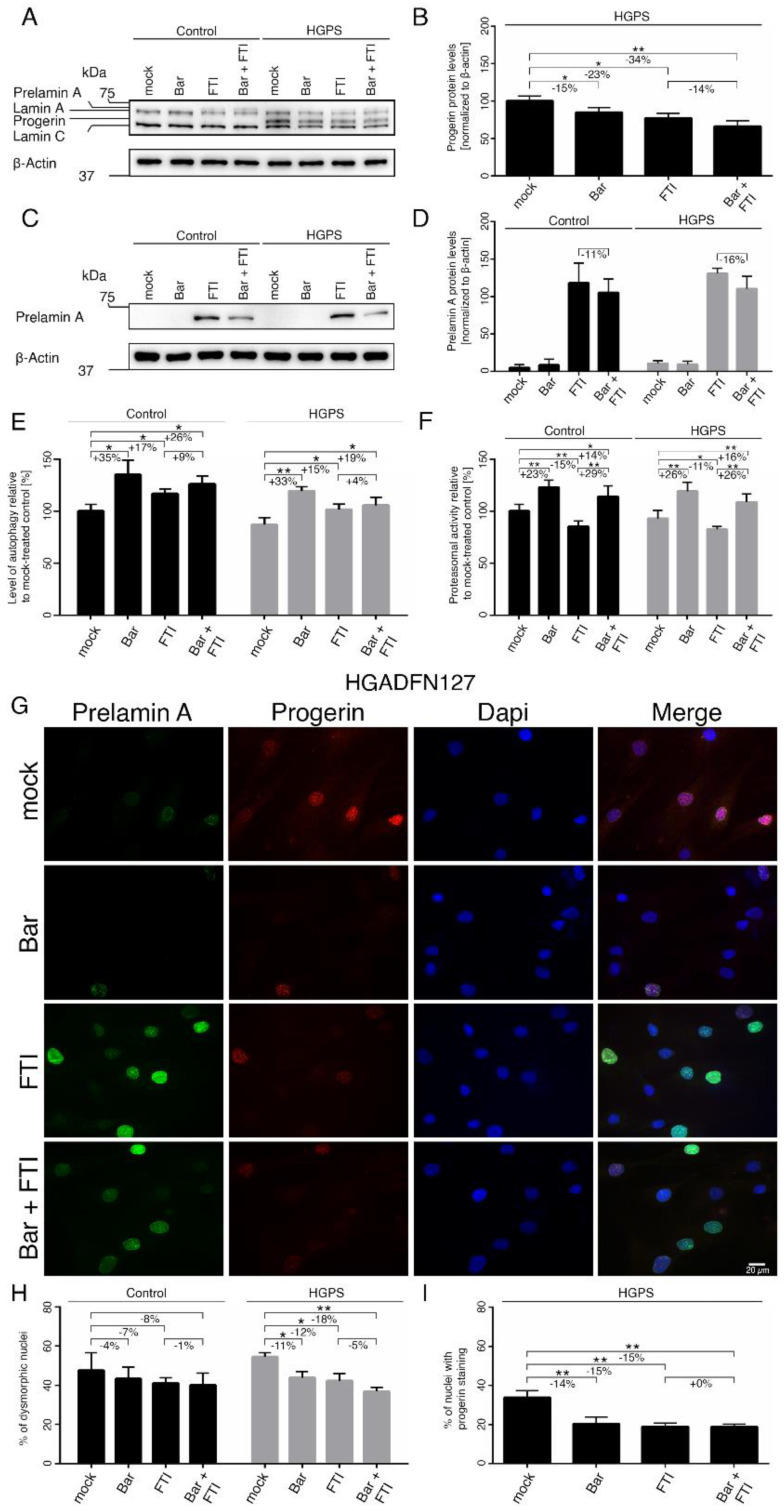
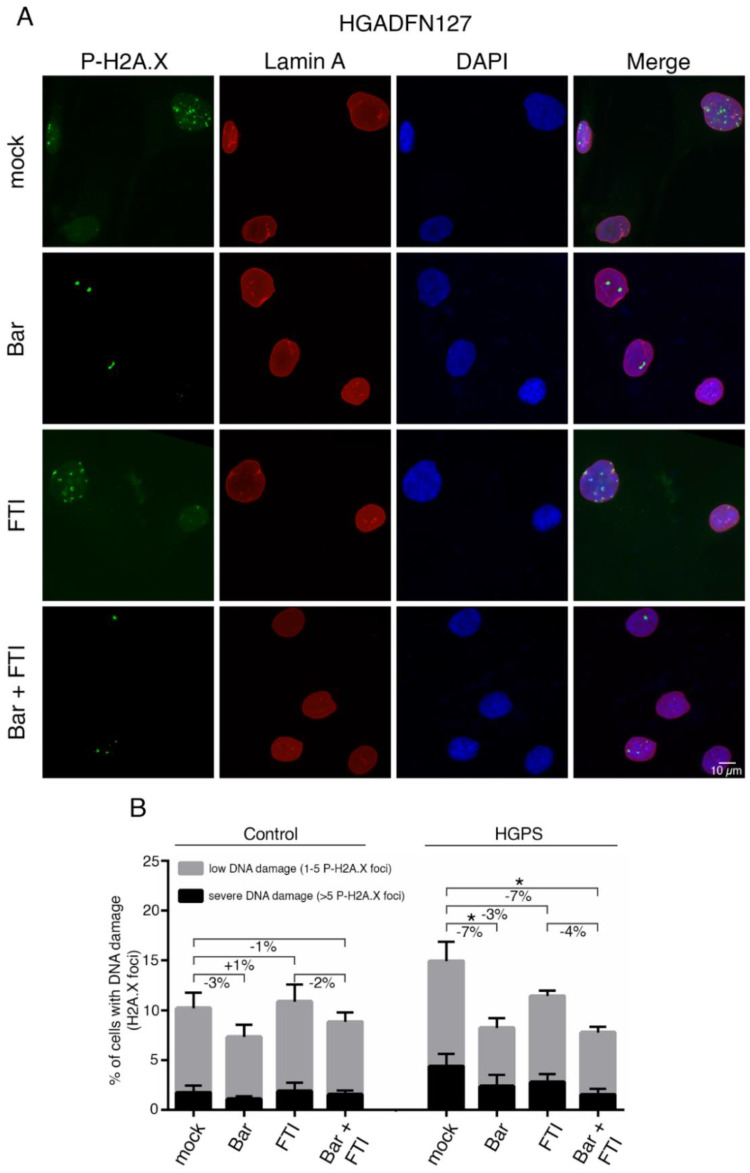
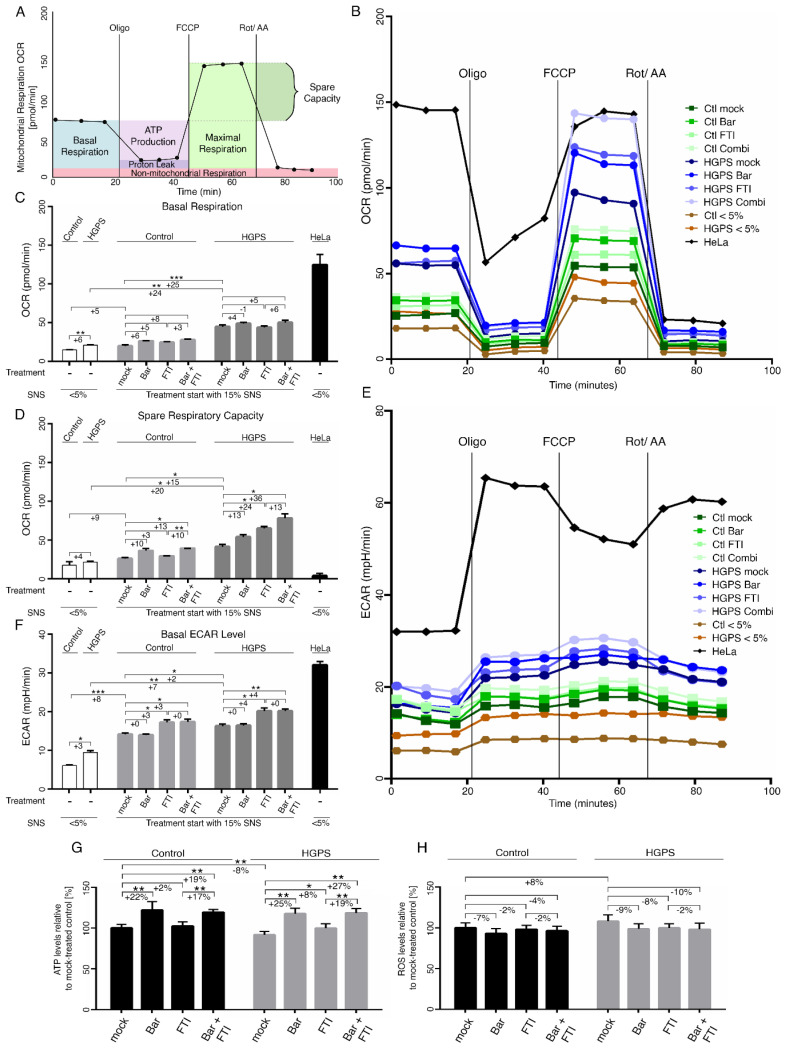
Hutchinson-Gilford progeria syndrome (HGPS) is an ultra-rare multisystem premature aging disorder that leads to early death (mean age of 14.7 years) due to myocardial infarction or stroke. Most cases have a de novo point mutation at position G608G within exon 11 of the LMNA gene. This mutation leads to the production of a permanently farnesylated truncated prelamin A protein called "progerin" that is toxic to the cells. Recently, farnesyltransferase inhibitor (FTI) lonafarnib has been approved by the FDA for the treatment of patients with HGPS. While lonafarnib treatment irrefutably ameliorates HGPS disease, it is however not a cure. FTI has been shown to cause several cellular side effects, including genomic instability as well as binucleated and donut-shaped nuclei. We report that, in addition to these cellular stresses, FTI caused an increased frequency of cytosolic DNA fragment formation. These extranuclear DNA fragments colocalized with cGAs and activated the cGAS-STING-STAT1 signaling axis, upregulating the expression of proinflammatory cytokines in FTI-treated human HGPS fibroblasts. Treatment with lonafarnib and baricitinib, a JAK-STAT inhibitor, not only prevented the activation of the cGAS STING-STAT1 pathway, but also improved the overall HGPS cellular homeostasis. These ameliorations included progerin levels, nuclear shape, proteostasis, cellular ATP, proliferation, and the reduction of cellular inflammation and senescence. Thus, we suggest that combining lonafarnib with baricitinib might provide an opportunity to reduce FTI cellular toxicity and ameliorate HGPS symptoms further than lonafarnib alone.
Keywords: JAK-STAT; age-related disease; baricitinib; farnesyltransferase inhibitor; inflammation; lamin; progeria; progerin; replicative senescence.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- Gordon L.B. PRF by the Numbers. The Progeria Research Foundation. [(accessed on 7 July 2021)]; Available online: https://www.progeriaresearch.org/prf-by-the-numbers/
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
