

Anti-Inflammatory Influences of Cystic Fibrosis Transmembrane Conductance Regulator Drugs on Lung Inflammation in Cystic Fibrosis
- PMID: 34299226
- PMCID: PMC8306345
- DOI: 10.3390/ijms22147606
Anti-Inflammatory Influences of Cystic Fibrosis Transmembrane Conductance Regulator Drugs on Lung Inflammation in Cystic Fibrosis
Abstract
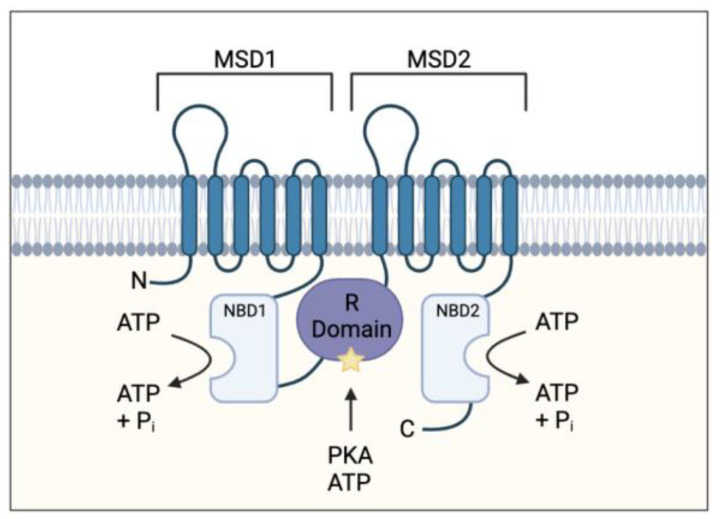
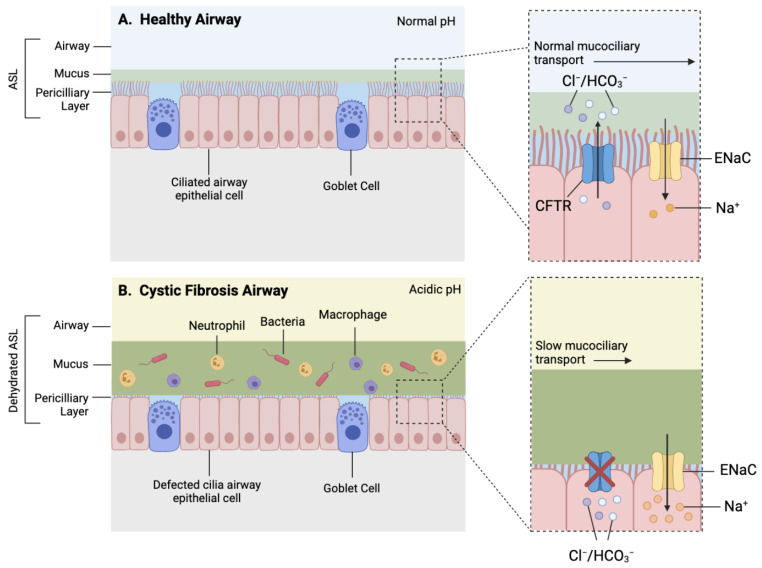
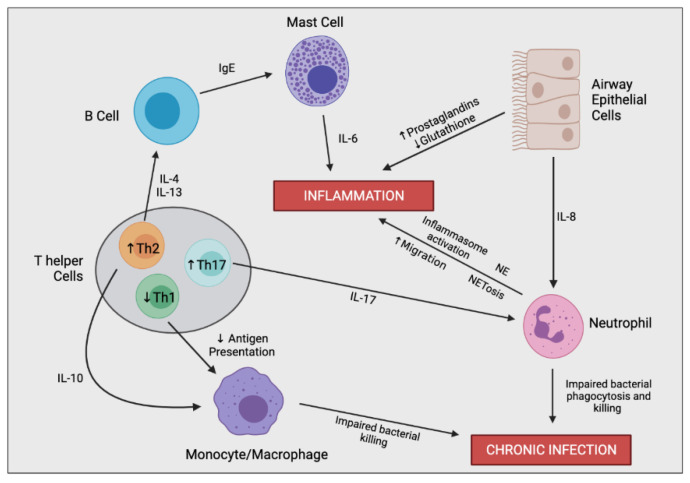
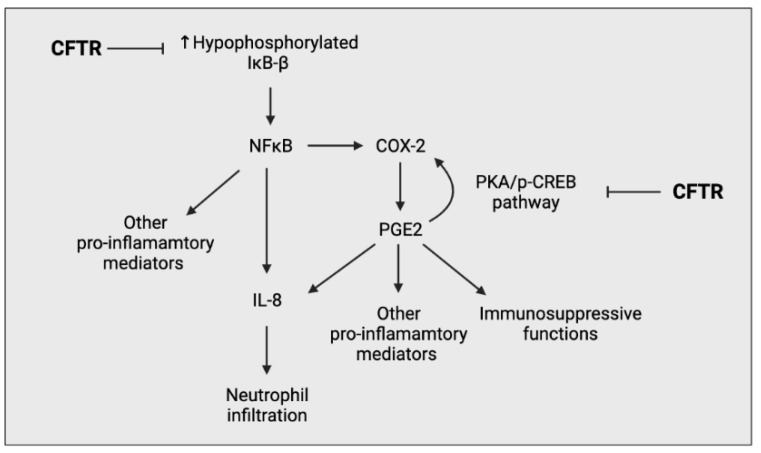
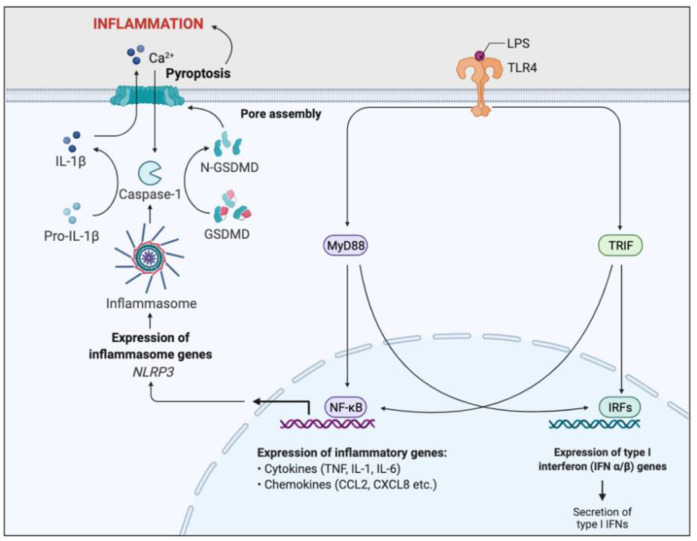
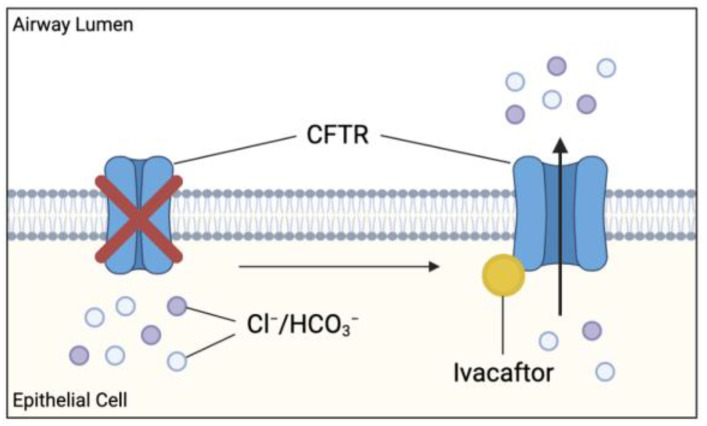
Cystic fibrosis (CF) is caused by a defect in the cystic fibrosis transmembrane conductance regulator protein (CFTR) which instigates a myriad of respiratory complications including increased vulnerability to lung infections and lung inflammation. The extensive influx of pro-inflammatory cells and production of mediators into the CF lung leading to lung tissue damage and increased susceptibility to microbial infections, creates a highly inflammatory environment. The CF inflammation is particularly driven by neutrophil infiltration, through the IL-23/17 pathway, and function, through NE, NETosis, and NLRP3-inflammasome formation. Better understanding of these pathways may uncover untapped therapeutic targets, potentially reducing disease burden experienced by CF patients. This review outlines the dysregulated lung inflammatory response in CF, explores the current understanding of CFTR modulators on lung inflammation, and provides context for their potential use as therapeutics for CF. Finally, we discuss the determinants that need to be taken into consideration to understand the exaggerated inflammatory response in the CF lung.
Keywords: CFTR modulator; anti-inflammatory treatment; cystic fibrosis; inflammation; ivacaftor; lumacaftor; lung inflammation.
Conflict of interest statement
The authors declare no conflict of interest. K.H.H., R.M.M., A.J. and E.K.S.-F. have nothing to disclose.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
