

Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease
- PMID: 34299236
- PMCID: PMC8307624
- DOI: 10.3390/ijms22147618
Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease
Abstract
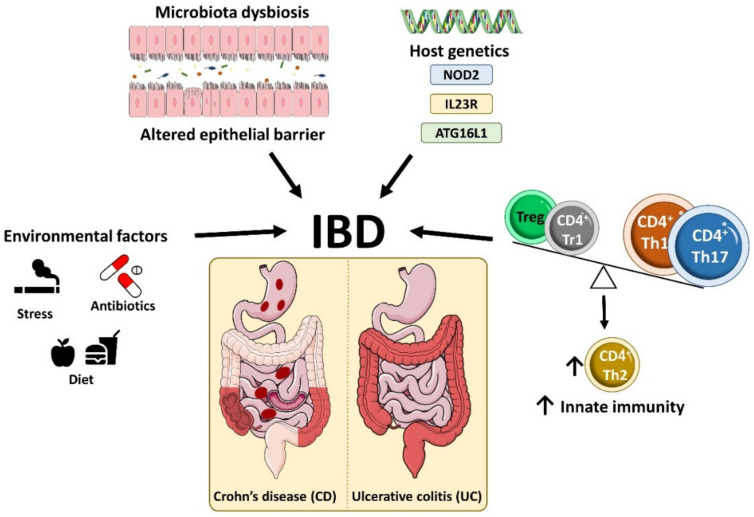
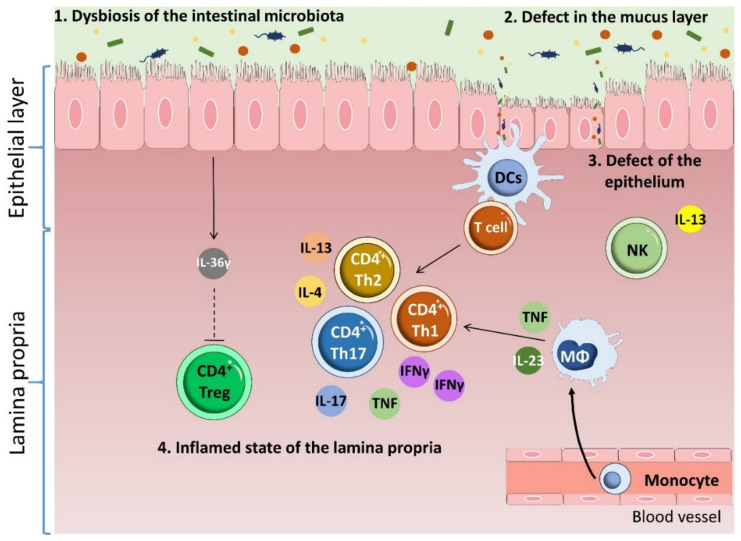
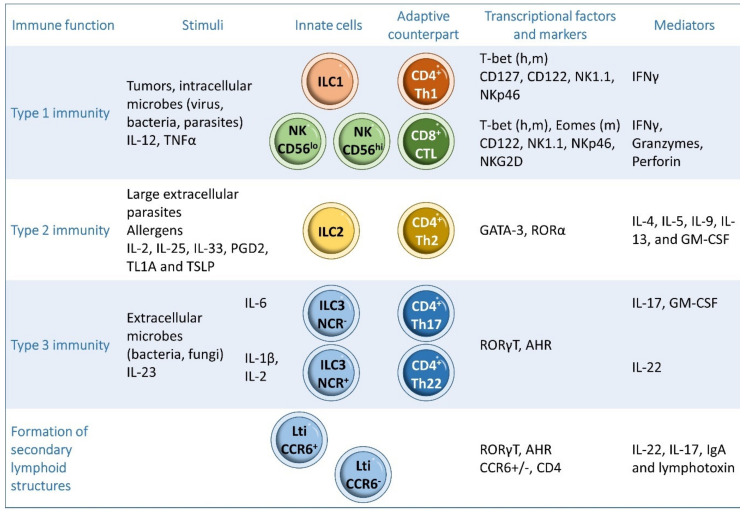
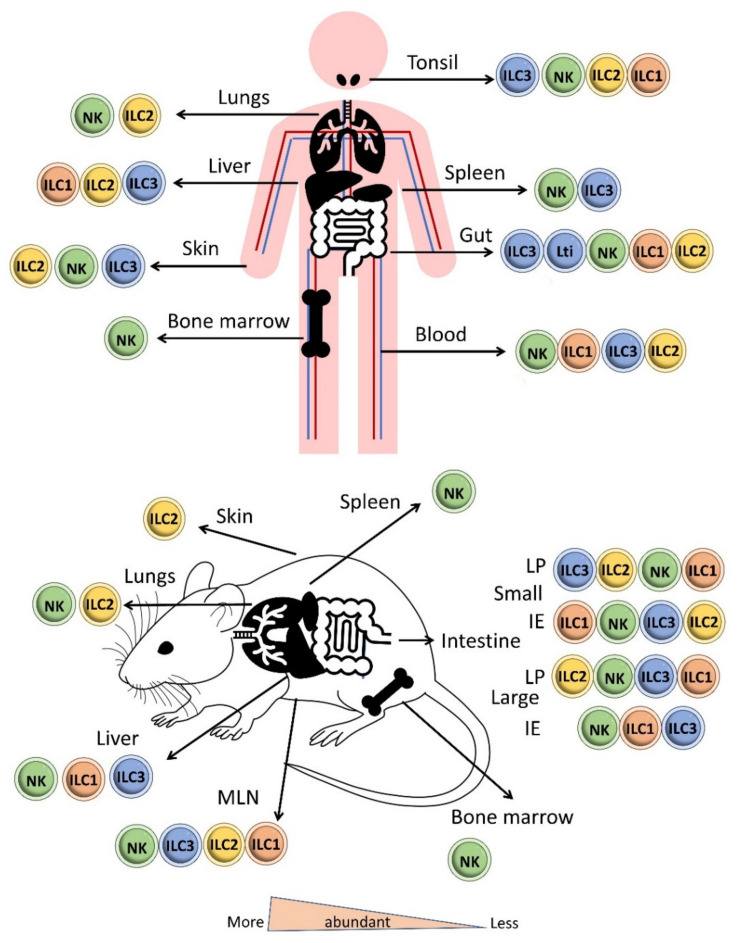
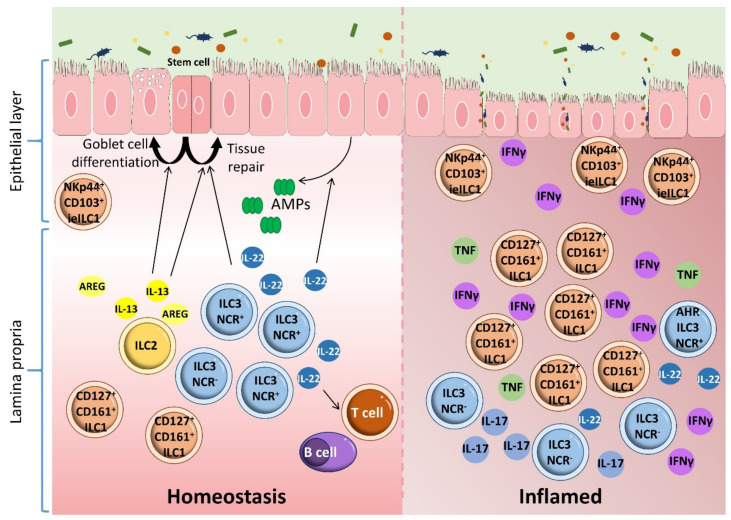
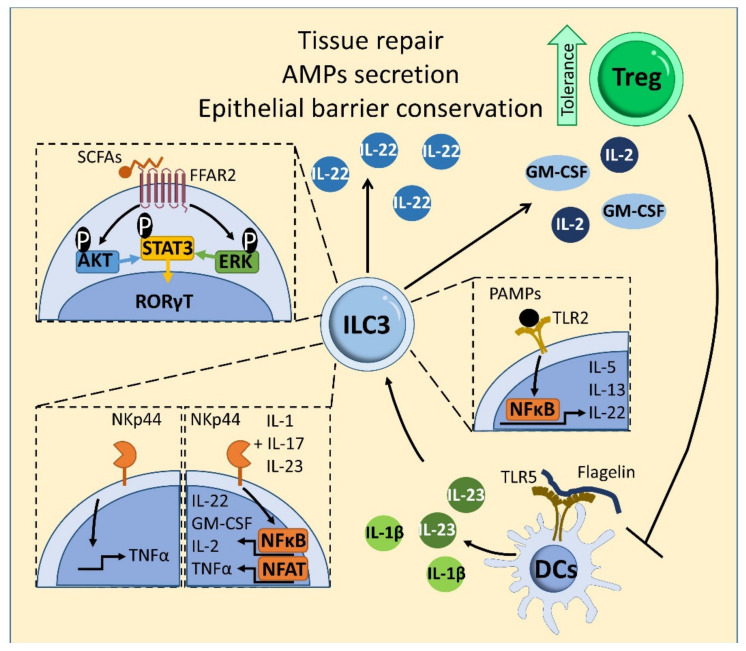
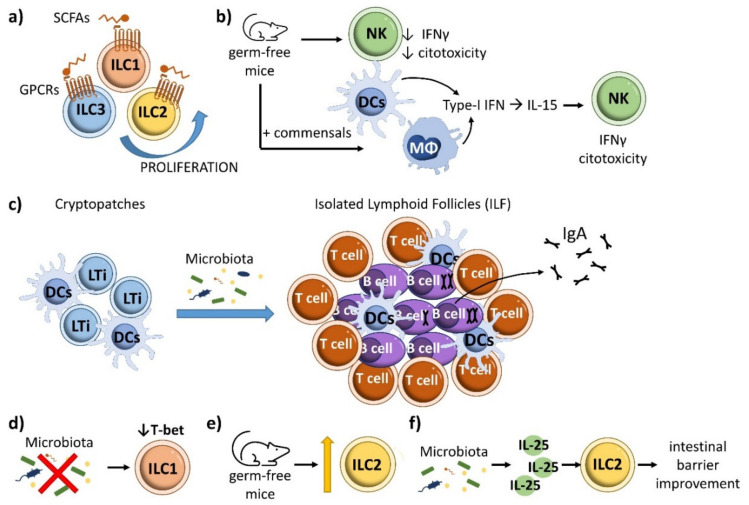
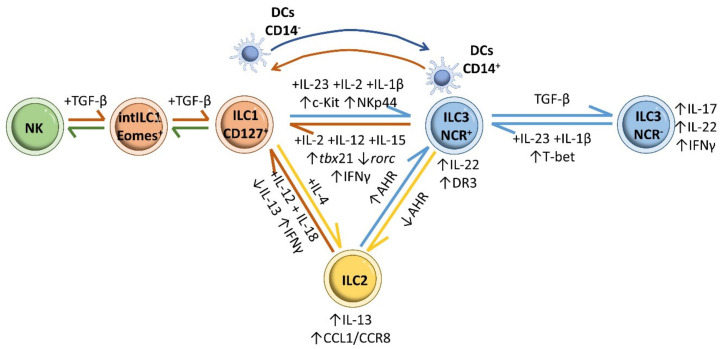
Inflammatory bowel disease (IBD) is a heterogeneous state of chronic intestinal inflammation of unknown cause encompassing Crohn's disease (CD) and ulcerative colitis (UC). IBD has been linked to genetic and environmental factors, microbiota dysbiosis, exacerbated innate and adaptive immunity and epithelial intestinal barrier dysfunction. IBD is classically associated with gut accumulation of proinflammatory Th1 and Th17 cells accompanied by insufficient Treg numbers and Tr1 immune suppression. Inflammatory T cells guide innate cells to perpetuate a constant hypersensitivity to microbial antigens, tissue injury and chronic intestinal inflammation. Recent studies of intestinal mucosal homeostasis and IBD suggest involvement of innate lymphoid cells (ILCs). These lymphoid-origin cells are innate counterparts of T cells but lack the antigen receptors expressed on B and T cells. ILCs play important roles in the first line of antimicrobial defense and contribute to organ development, tissue protection and regeneration, and mucosal homeostasis by maintaining the balance between antipathogen immunity and commensal tolerance. Intestinal homeostasis requires strict regulation of the quantity and activity of local ILC subpopulations. Recent studies demonstrated that changes to ILCs during IBD contribute to disease development. A better understanding of ILC behavior in gastrointestinal homeostasis and inflammation will provide valuable insights into new approaches to IBD treatment. This review summarizes recent research into ILCs in intestinal homeostasis and the latest advances in the understanding of the role of ILCs in IBD, with particular emphasis on the interaction between microbiota and ILC populations and functions.
Keywords: inflammatory bowel disease; innate lymphoid cells; intestinal homeostasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Global burden of inflammatory bowel disease 2017 Collaborators The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020;5:17–30. doi: 10.1016/S2468-1253(19)30333-4. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
