

The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation
- PMID: 34300201
- PMCID: PMC8307963
- DOI: 10.3390/jcm10143034
The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation
Abstract
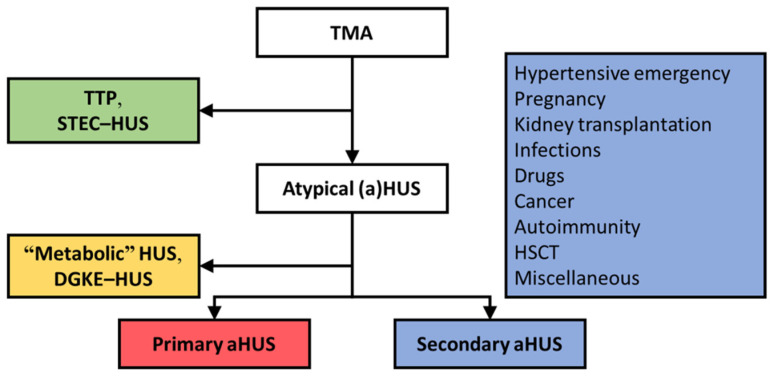
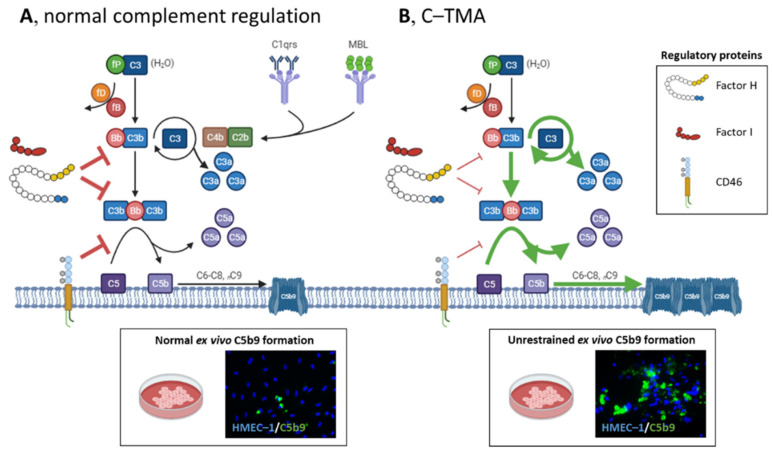
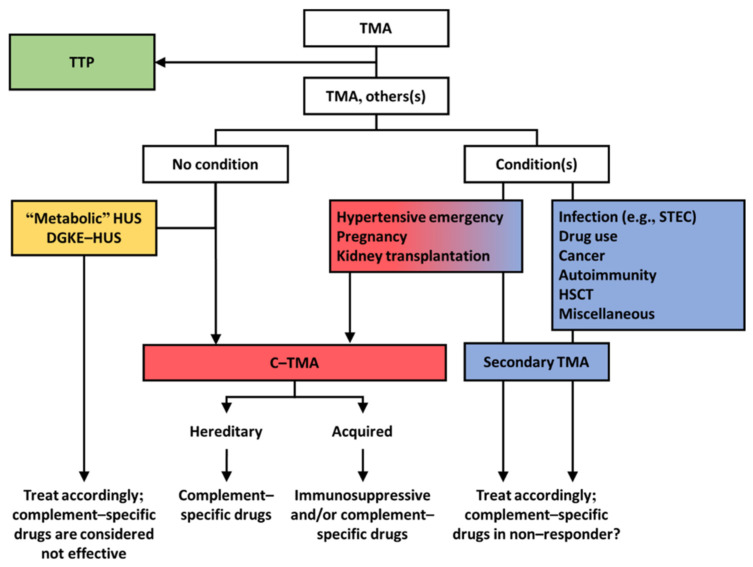
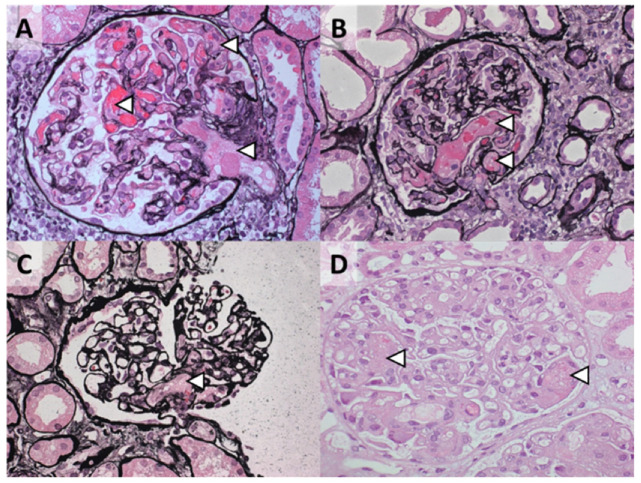
Thrombotic microangiopathy (TMA) is a rare and potentially life-threatening condition that can be caused by a heterogeneous group of diseases, often affecting the brain and kidneys. TMAs should be classified according to etiology to indicate targets for treatment. Complement dysregulation is an important cause of TMA that defines cases not related to coexisting conditions, that is, primary atypical hemolytic uremic syndrome (HUS). Ever since the approval of therapeutic complement inhibition, the approach of TMA has focused on the recognition of primary atypical HUS. Recent advances, however, demonstrated the pivotal role of complement dysregulation in specific subtypes of patients considered to have secondary atypical HUS. This is particularly the case in patients presenting with coexisting hypertensive emergency, pregnancy, and kidney transplantation, shifting the paradigm of disease. In contrast, complement dysregulation is uncommon in patients with other coexisting conditions, such as bacterial infection, drug use, cancer, and autoimmunity, among other disorders. In this review, we performed a critical appraisal on complement dysregulation and the use of therapeutic complement inhibition in TMAs associated with coexisting conditions and outline a pragmatic approach to diagnosis and treatment. For future studies, we advocate the term complement-mediated TMA as opposed to the traditional atypical HUS-type classification.
Keywords: atypical hemolytic uremic syndrome; complement; eculizumab; hypertensive emergency; kidney transplantation; pregnancy; thrombotic microangiopathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Loirat C., Fakhouri F., Ariceta G., Besbas N., Bitzan M., Bjerre A., Coppo R., Emma F., Johnson S., Karpman D., et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016;31:15–39. doi: 10.1007/s00467-015-3076-8. - DOI - PubMed
-
- Goodship T.H., Cook H.T., Fakhouri F., Fervenza F.C., Fremeaux-Bacchi V., Kavanagh D., Nester C.M., Noris M., Pickering M.C., Rodriguez de Cordoba S., et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91:539–551. doi: 10.1016/j.kint.2016.10.005. - DOI - PubMed
-
- Furlan M., Robles R., Galbusera M., Remuzzi G., Kyrle P.A., Brenner B., Krause M., Scharrer I., Aumann V., Mittler U., et al. Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N. Engl. J. Med. 1998;339:1578–1584. doi: 10.1056/NEJM199811263392202. - DOI - PubMed
-
- Noris M., Caprioli J., Bresin E., Mossali C., Pianetti G., Gamba S., Daina E., Fenili C., Castelletti F., Sorosina A., et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. 2010;5:1844–1859. doi: 10.2215/CJN.02210310. - DOI - PMC - PubMed
-
- Fremeaux-Bacchi V., Fakhouri F., Garnier A., Bienaime F., Dragon-Durey M.A., Ngo S., Moulin B., Servais A., Provot F., Rostaing L., et al. Genetics and outcome of atypical hemolytic uremic syndrome: A nationwide French series comparing children and adults. Clin. J. Am. Soc. Nephrol. 2013;8:554–562. doi: 10.2215/CJN.04760512. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
