

Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing
- PMID: 34301788
- PMCID: PMC8783930
- DOI: 10.1158/2159-8290.CD-20-1631
Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing
Abstract
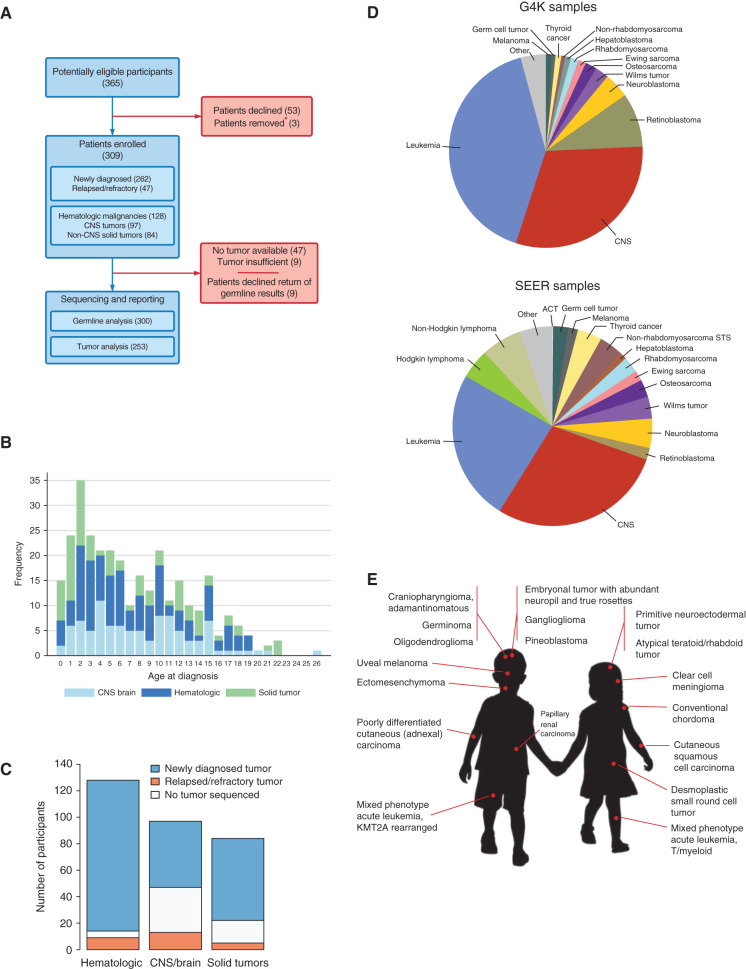
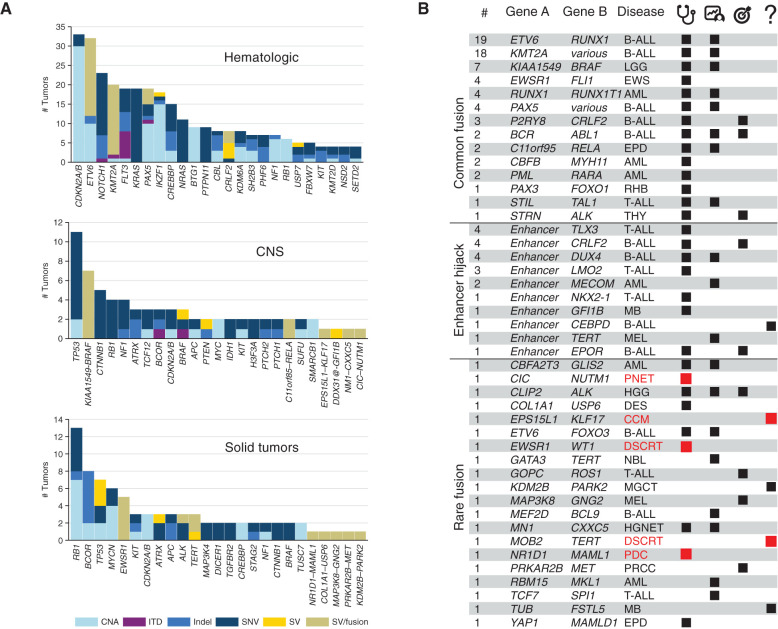
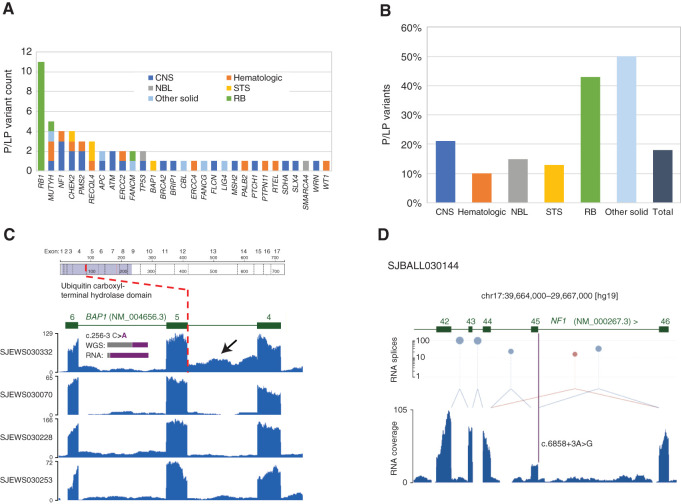
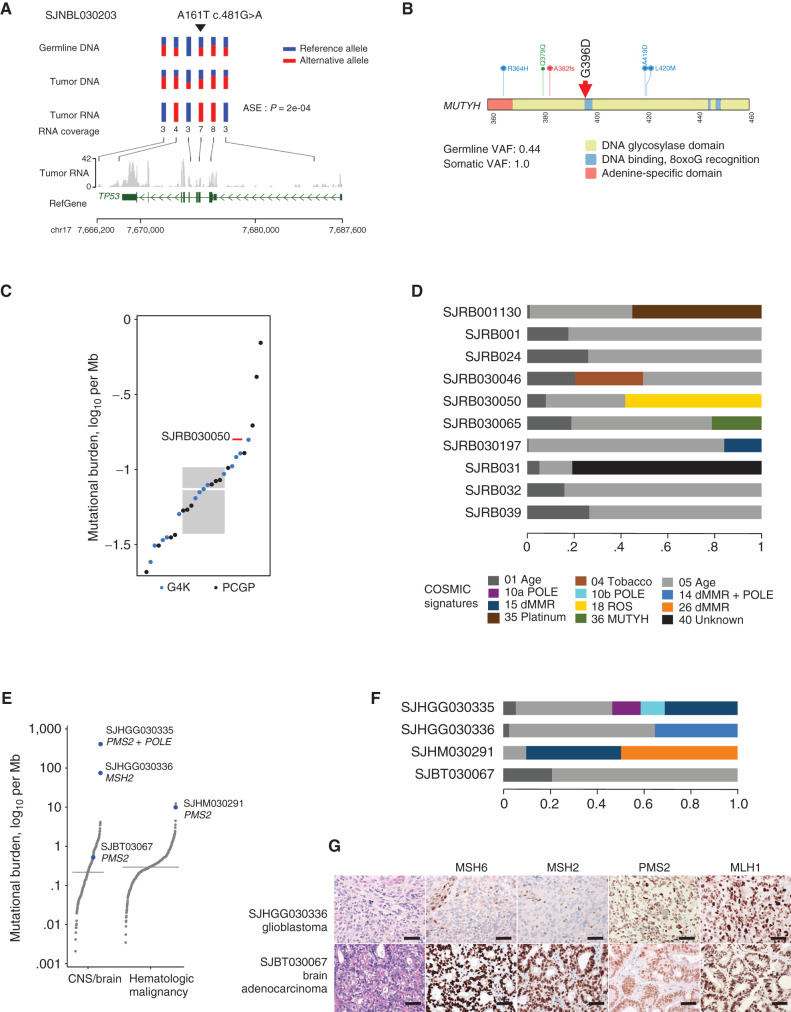
Genomic studies of pediatric cancer have primarily focused on specific tumor types or high-risk disease. Here, we used a three-platform sequencing approach, including whole-genome sequencing (WGS), whole-exome sequencing (WES), and RNA sequencing (RNA-seq), to examine tumor and germline genomes from 309 prospectively identified children with newly diagnosed (85%) or relapsed/refractory (15%) cancers, unselected for tumor type. Eighty-six percent of patients harbored diagnostic (53%), prognostic (57%), therapeutically relevant (25%), and/or cancer-predisposing (18%) variants. Inclusion of WGS enabled detection of activating gene fusions and enhancer hijacks (36% and 8% of tumors, respectively), small intragenic deletions (15% of tumors), and mutational signatures revealing of pathogenic variant effects. Evaluation of paired tumor-normal data revealed relevance to tumor development for 55% of pathogenic germline variants. This study demonstrates the power of a three-platform approach that incorporates WGS to interrogate and interpret the full range of genomic variants across newly diagnosed as well as relapsed/refractory pediatric cancers.
Significance: Pediatric cancers are driven by diverse genomic lesions, and sequencing has proven useful in evaluating high-risk and relapsed/refractory cases. We show that combined WGS, WES, and RNA-seq of tumor and paired normal tissues enables identification and characterization of genetic drivers across the full spectrum of pediatric cancers. This article is highlighted in the In This Issue feature, p. 2945.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
![Figure 3. Using multi-omics data to interpret pathogenicity of SVs. A, GenomePaint plots showing two regions, chr8:128500000–130600000 (top) and chr5: 170710000–170790000 (bottom) [hg19], from SJTALL030071. Both panels consist of the RefSeq gene model in green, with MYC and TLX3 highlighted by red boxes; orange bars show regions of copy-number gain supported by increased WGS coverage plotted as the blue histogram immediately below. Gray lollipops marked t(5;8) indicate the position of the translocation breakpoint. RNA-seq coverage is shown below the whole-genome coverage histogram. Additional data from NCI-TARGET are also shown, with narrow red bars representing regions of copy-number gain and gray lollipops representing SV breakpoints surrounding the TLX3 locus. A region or recurrent copy-number gain in TARGET samples is adjacent to the chromosome 8 breakpoint in SJTALL030071. Generally high but nonspecific RNA-seq coverage at this locus suggests a region of high transcriptional activity (distal MYC enhancer) is brought into proximity of TLX3 by the translocation. CNV, copy-number variation. B, A rank-order plot of T-ALL from TARGET showing expression levels of TLX3 mRNA in a set of TLX3-activated tumors compared with tumors in which TLX3 was not activated. SJTALL030071 TLX3 expression (red dot) groups with the activated set. C, Allele-specific expression (ASE) of the TLX3 locus in SJTALL030071. The tumor DNA (top row) shows a series of heterozygous alleles in the TLX3 locus (blue and red stacked bars show relative VAF from WGS data). In the RNA-seq data (second row), expression of only one allele is observed. RNA coverage, read counts at each allele, are shown numerically (third row). Beneath the read counts, black lines map the locations of the alleles to the chromosome 5 coordinates surrounding the TLX3 locus. Beneath the coordinate line, the location of the SJTALL030071 translocation breakpoint is indicated. D, Two-dimensional t-SNE plot of RNA-seq–derived gene expression data from 264 T-ALL samples (41). Major T-ALL subgroups are indicated on the plot, with SJTALL030071 localizing among the TLX3 cluster, as shown by the black arrow. E, Schematic representation of an ETV6–FOXO3 fusion found in SJBALL030052 joining the N-terminal region to the ETV6 sterile alpha motif domain (green) with oligomerization interfaces (red) and ETS domain (purple), with the C-terminal FOXO3 forkhead binding (green), KIX binding (purple), and transactivation domains (red). F, Two-dimensional t-SNE plot of RNA-seq–derived gene expression data from 1,988 B-ALL samples (42). Major subgroups are indicated on the plot, with SJBALL030052 localizing to the periphery of the ETV6–RUNX1 subgroup, as shown by the black arrow.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/2faf/9414289/90708c402e4a/3008fig3.jpg)

Comment in
- doi: 10.1158/2159-8290.CD-11-12-ITI
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
