

Strainberry: automated strain separation in low-complexity metagenomes using long reads
- PMID: 34301928
- PMCID: PMC8302730
- DOI: 10.1038/s41467-021-24515-9
Strainberry: automated strain separation in low-complexity metagenomes using long reads
Abstract
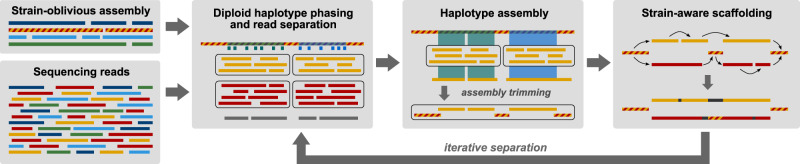
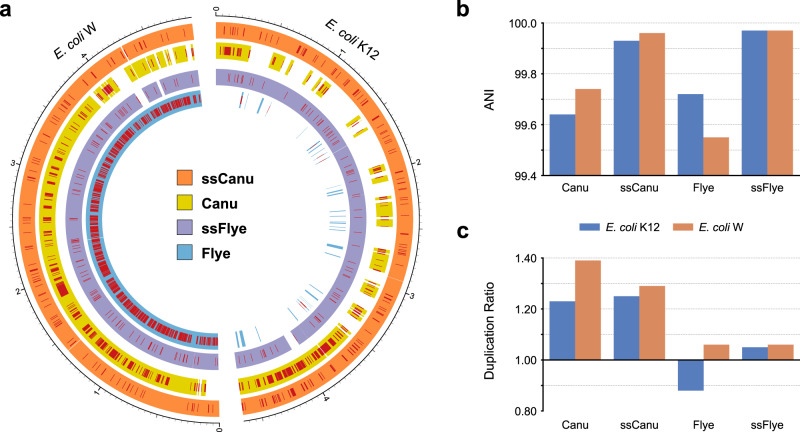
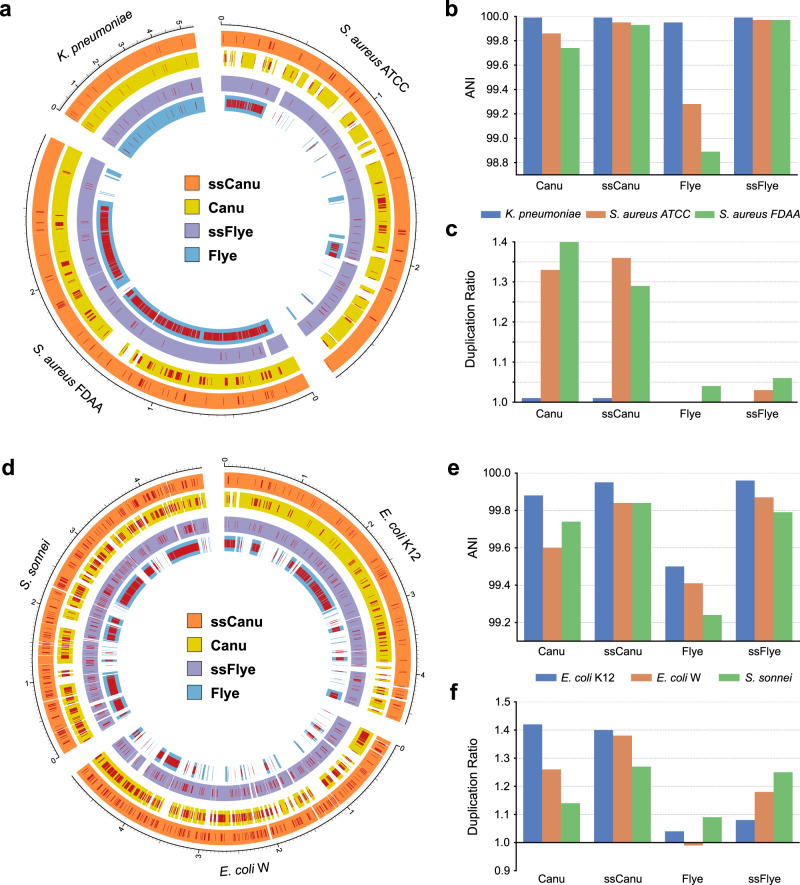
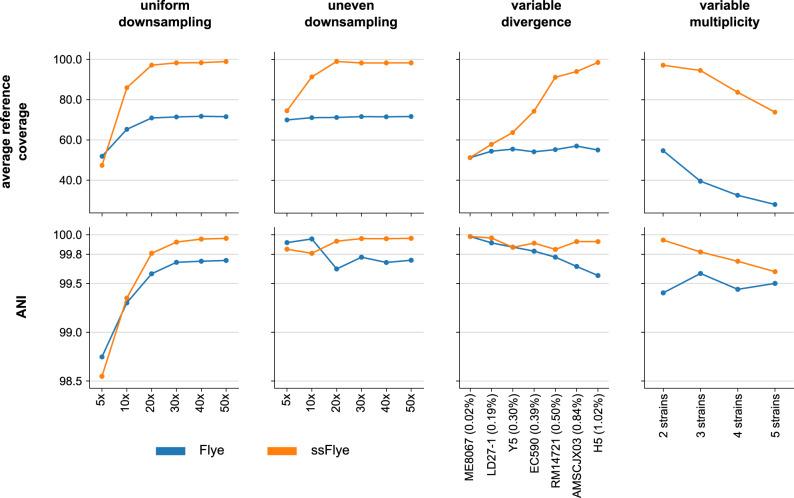
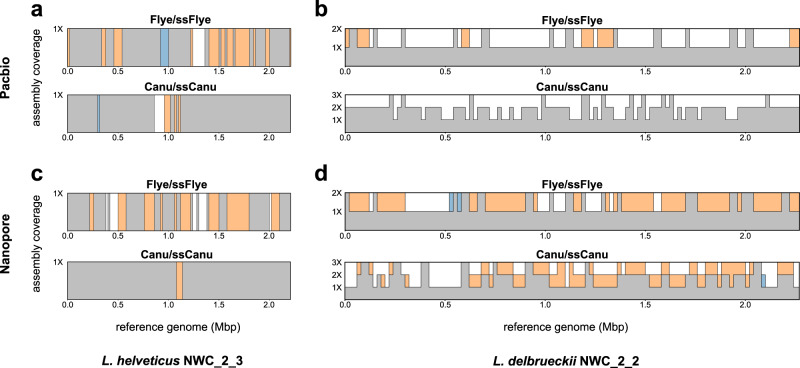
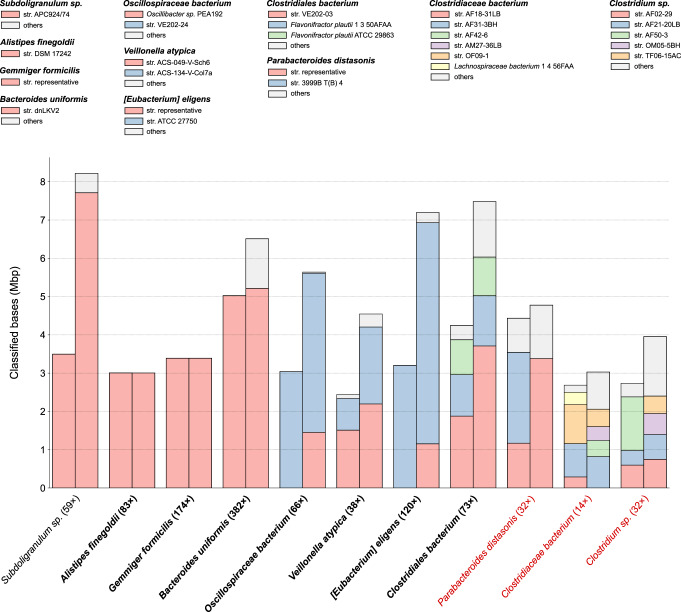
High-throughput short-read metagenomics has enabled large-scale species-level analysis and functional characterization of microbial communities. Microbiomes often contain multiple strains of the same species, and different strains have been shown to have important differences in their functional roles. Recent advances on long-read based methods enabled accurate assembly of bacterial genomes from complex microbiomes and an as-yet-unrealized opportunity to resolve strains. Here we present Strainberry, a metagenome assembly pipeline that performs strain separation in single-sample low-complexity metagenomes and that relies uniquely on long-read data. We benchmarked Strainberry on mock communities for which it produces strain-resolved assemblies with near-complete reference coverage and 99.9% base accuracy. We also applied Strainberry on real datasets for which it improved assemblies generating 20-118% additional genomic material than conventional metagenome assemblies on individual strain genomes. We show that Strainberry is also able to refine microbial diversity in a complex microbiome, with complete separation of strain genomes. We anticipate this work to be a starting point for further methodological improvements on strain-resolved metagenome assembly in environments of higher complexities.
© 2021. The Author(s).
Conflict of interest statement
A.E.D. holds equity in and is cofounder and CSO of Longas Technologies Pty Ltd, which is a commercial entity developing synthetic long-read sequencing technologies. The authors declare no additional competing interest.
Figures
References
-
- Blaser MJ, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
