

Trajectory of Growth of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants in Houston, Texas, January through May 2021, Based on 12,476 Genome Sequences
- PMID: 34303698
- PMCID: PMC8299152
- DOI: 10.1016/j.ajpath.2021.07.002
Trajectory of Growth of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants in Houston, Texas, January through May 2021, Based on 12,476 Genome Sequences
Abstract
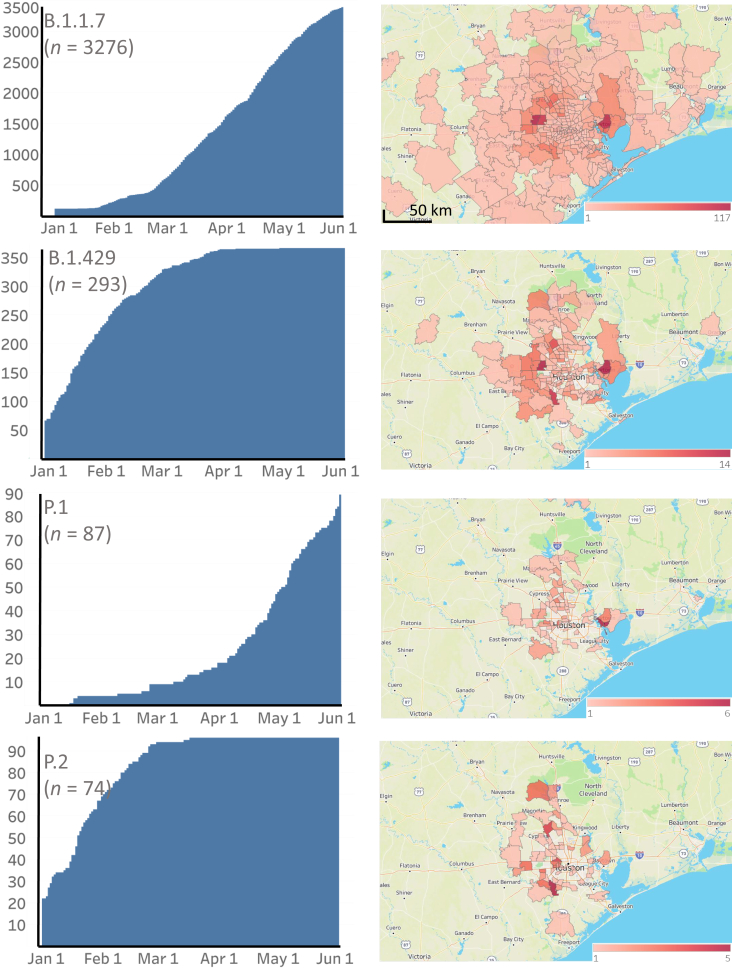
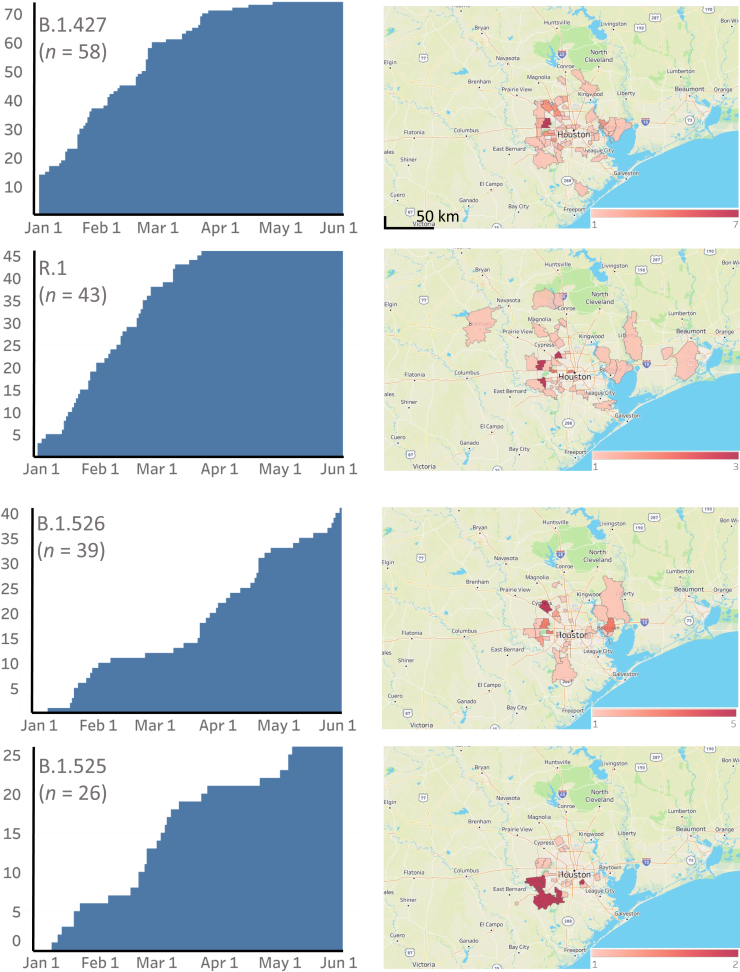
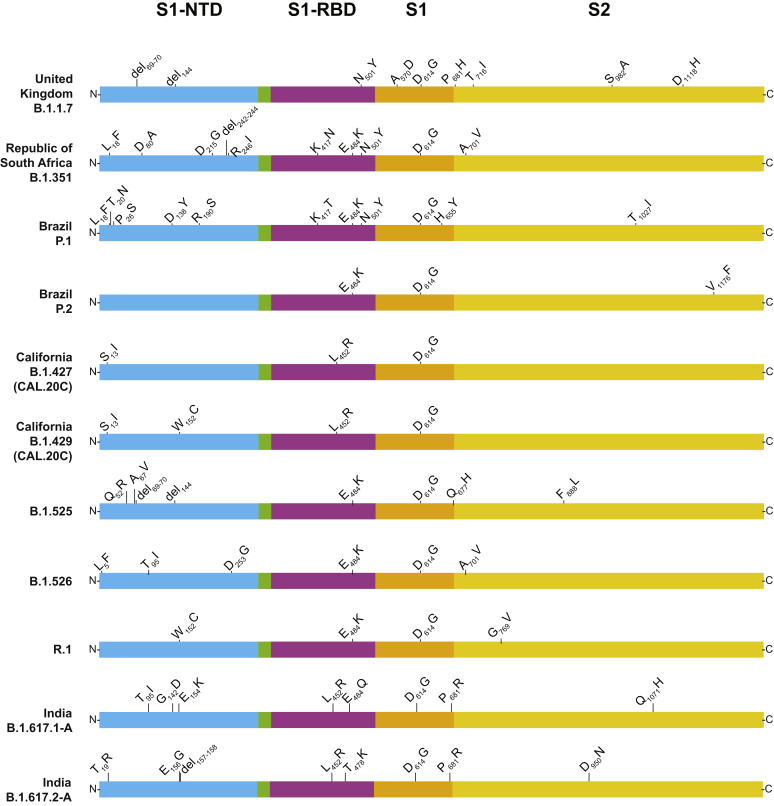
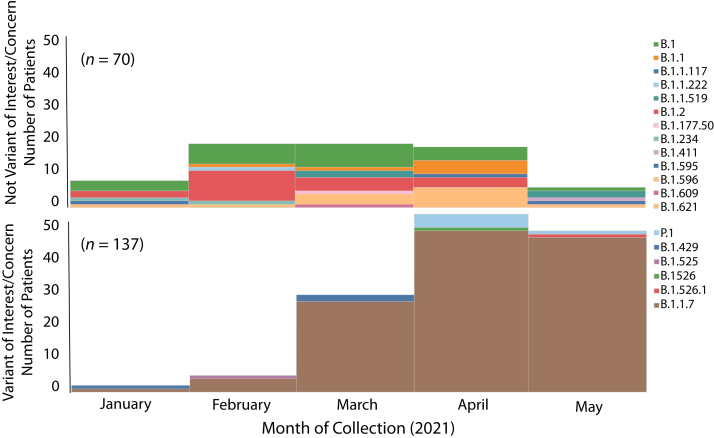
Certain genetic variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are of substantial concern because they may be more transmissible or detrimentally alter the pandemic course and disease features in individual patients. SARS-CoV-2 genome sequences from 12,476 patients in the Houston Methodist health care system diagnosed from January 1 through May 31, 2021 are reported here. Prevalence of the B.1.1.7 (Alpha) variant increased rapidly and caused 63% to 90% of new cases in the latter half of May. Eleven B.1.1.7 genomes had an E484K replacement in spike protein, a change also identified in other SARS-CoV-2 lineages. Compared with non-B.1.1.7-infected patients, individuals with B.1.1.7 had a significantly lower cycle threshold (a proxy for higher virus load) and significantly higher hospitalization rate. Other variants [eg, B.1.429 and B.1.427 (Epsilon), P.1 (Gamma), P.2 (Zeta), and R.1] also increased rapidly, although the magnitude was less than that in B.1.1.7. Twenty-two patients infected with B.1.617.1 (Kappa) or B.1.617.2 (Delta) variants had a high rate of hospitalization. Breakthrough cases (n = 207) in fully vaccinated patients were caused by a heterogeneous array of virus genotypes, including many not currently designated variants of interest or concern. In the aggregate, this study delineates the trajectory of SARS-CoV-2 variants circulating in a major metropolitan area, documents B.1.1.7 as the major cause of new cases in Houston, TX, and heralds the arrival of B.1.617 variants in the metroplex.
Copyright © 2021 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures




References
-
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. - PMC - PubMed
-
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W., China Novel Coronavirus I., Research T. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. - PMC - PubMed
-
- Chan J.F., Yuan S., Kok K.H., To K.K., Chu H., Yang J., Xing F., Liu J., Yip C.C., Poon R.W., Tsoi H.W., Lo S.K., Chan K.H., Poon V.K., Chan W.M., Ip J.D., Cai J.P., Cheng V.C., Chen H., Hui C.K., Yuen K.Y. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
