

Metabolic and Immunological Subtypes of Esophageal Cancer Reveal Potential Therapeutic Opportunities
- PMID: 34307352
- PMCID: PMC8295652
- DOI: 10.3389/fcell.2021.667852
Metabolic and Immunological Subtypes of Esophageal Cancer Reveal Potential Therapeutic Opportunities
Abstract
Background: Esophageal cancer has the sixth highest rate of cancer-associated deaths worldwide, with many patients displaying metastases and chemotherapy resistance. We sought to find subtypes to see if precision medicine could play a role in finding new potential targets and predicting responses to therapy. Since metabolism not only drives cancers but also serves as a readout, metabolism was examined as a key reporter for differences.
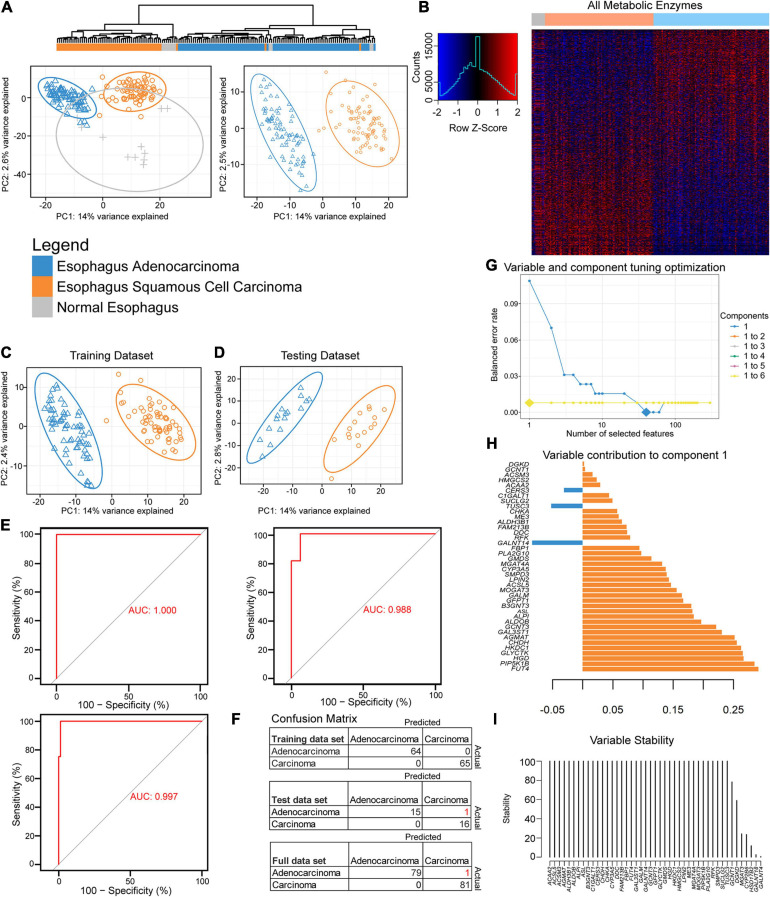
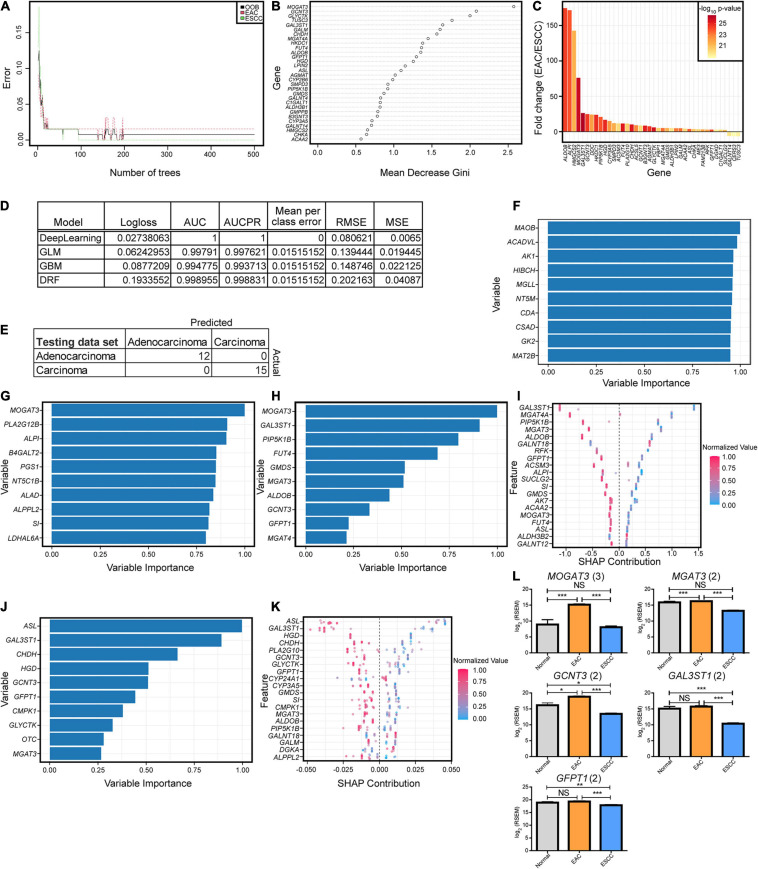
Methods: Unsupervised and supervised classification methods, including hierarchical clustering, partial least squares discriminant analysis, k-nearest neighbors, and machine learning techniques, were used to discover and display two major subgroups. Genes, pathways, gene ontologies, survival, and immune differences between the groups were further examined, along with biomarkers between the groups and against normal tissue.
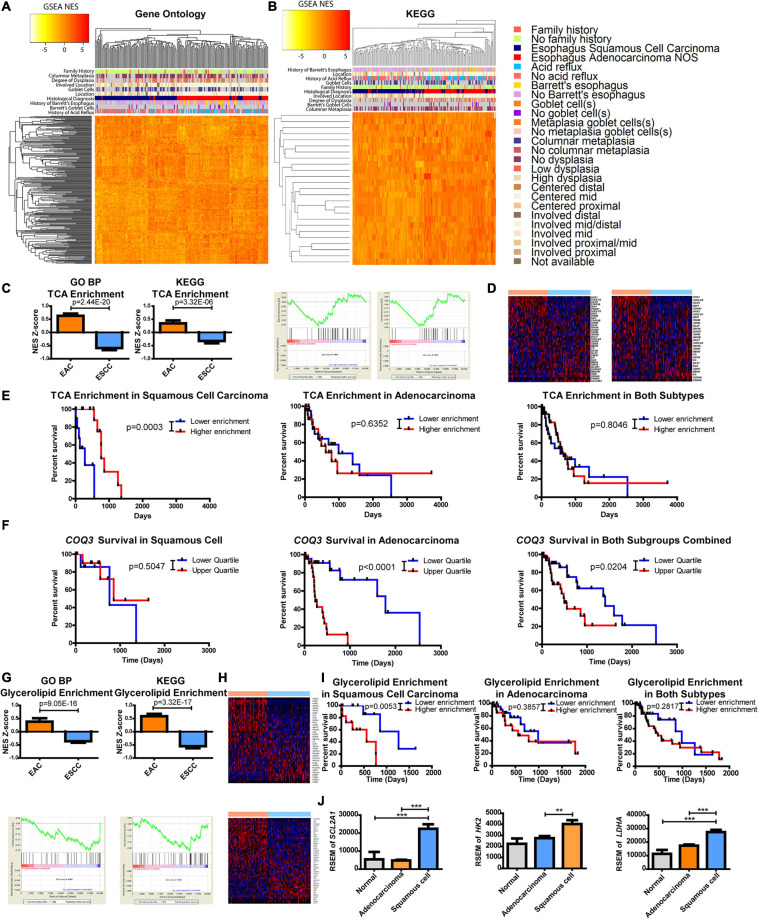
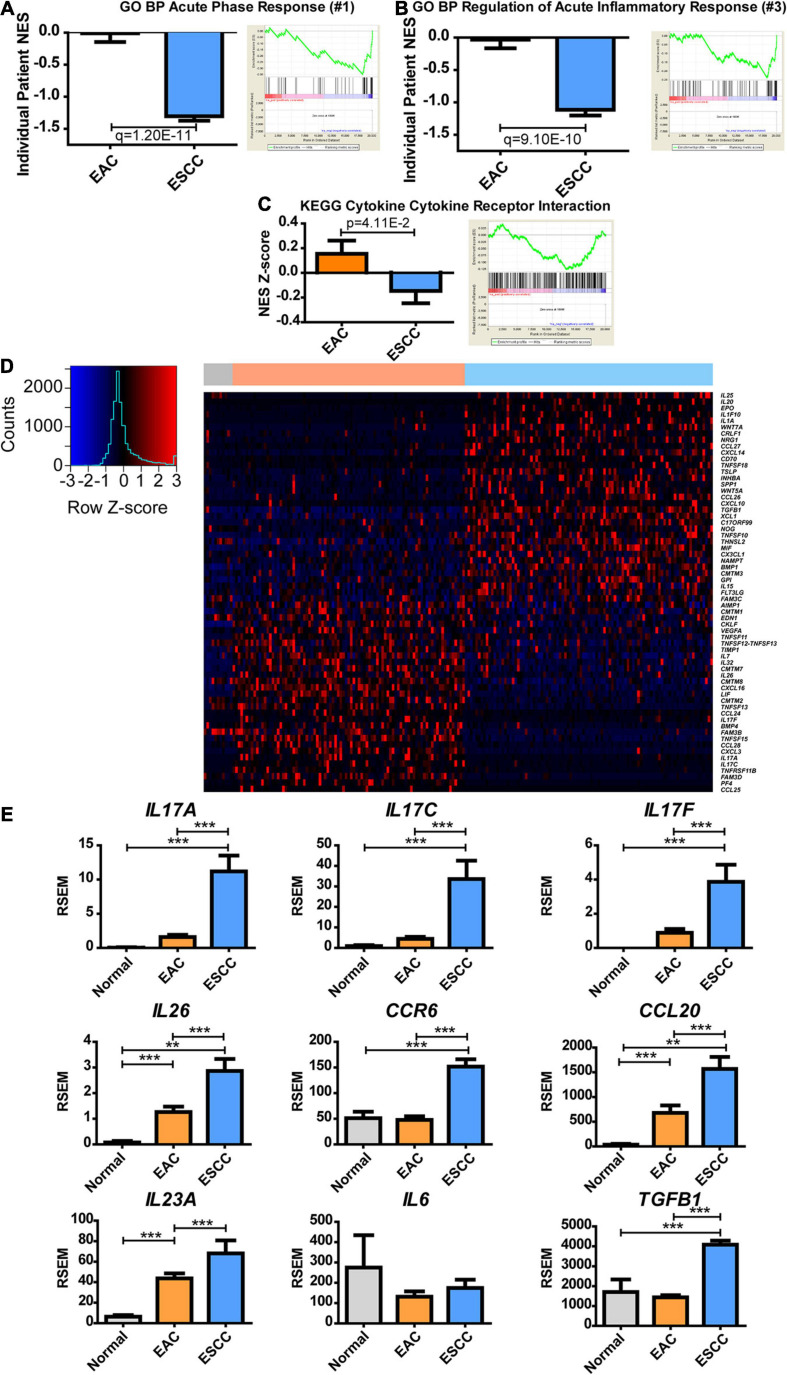
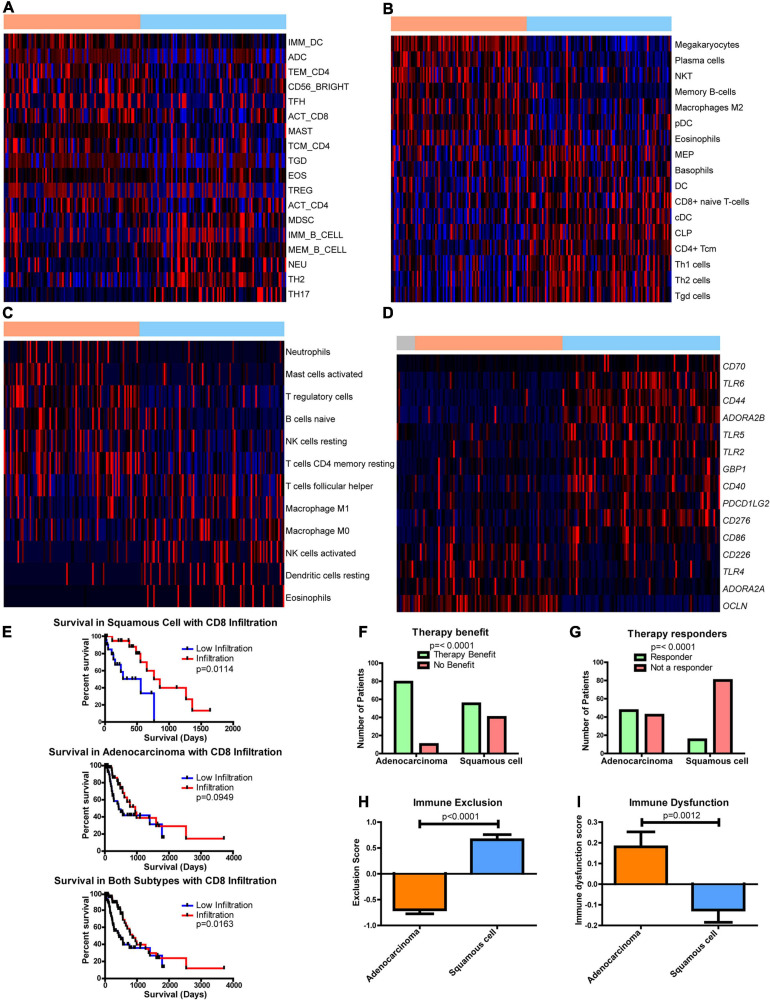
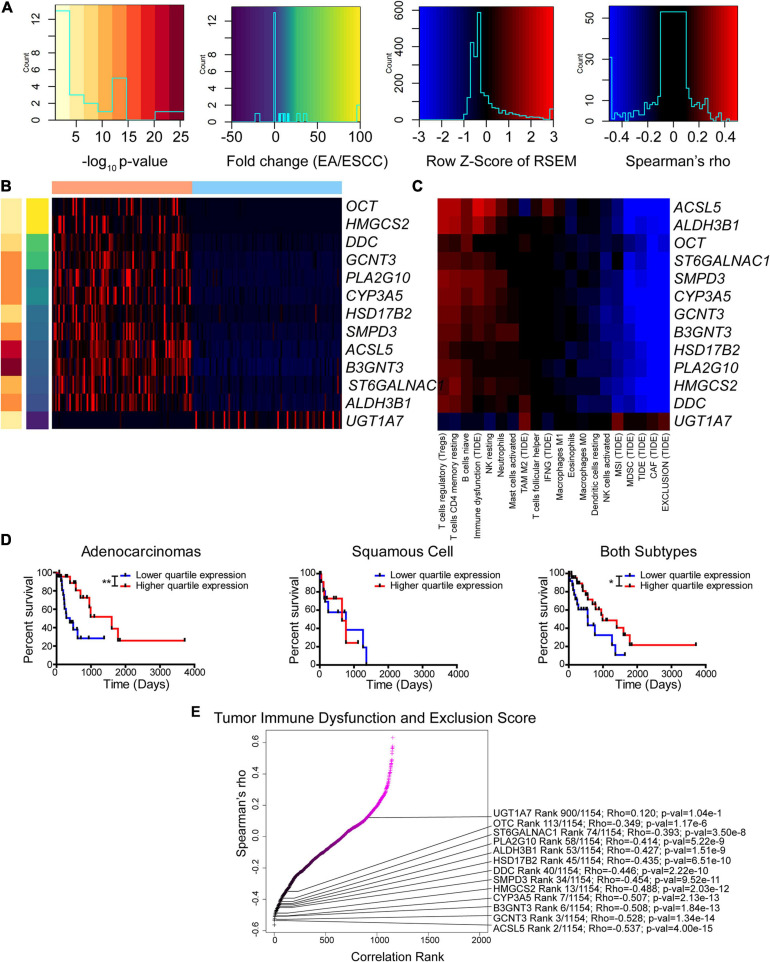
Results: Esophageal cancer had two major unique metabolic profiles observed between the histological subtypes esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC). The metabolic differences suggest that ESCC depends on glycolysis, whereas EAC relies more on oxidative metabolism, catabolism of glycolipids, the tricarboxylic acid (TCA) cycle, and the electron transport chain. We also noted a robust prognostic risk associated with COQ3 expression. In addition to the metabolic alterations, we noted significant alterations in key pathways regulating immunity, including alterations in cytokines and predicted immune infiltration. ESCC appears to have increased signature associated with dendritic cells, Th17, and CD8 T cells, the latter of which correlate with survival in ESCC. We bioinformatically observed that ESCC may be more responsive to checkpoint inhibitor therapy than EAC and postulate targets to enhance therapy further. Lastly, we highlight correlations between differentially expressed enzymes and the potential immune status.
Conclusion: Overall, these results highlight the extreme differences observed between the histological subtypes and may lead to novel biomarkers, therapeutic strategies, and differences in therapeutic response for targeting each esophageal cancer subtype.
Keywords: biomarkers; cancer metabolism; esophageal cancer; immunological subtypes; metabolic subtypes.
Copyright © 2021 King, Qiu, Yu and Singh.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials