

STRONG: metagenomics strain resolution on assembly graphs
- PMID: 34311761
- PMCID: PMC8311964
- DOI: 10.1186/s13059-021-02419-7
STRONG: metagenomics strain resolution on assembly graphs
Abstract
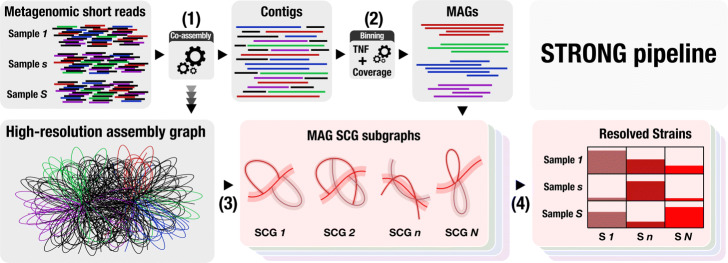
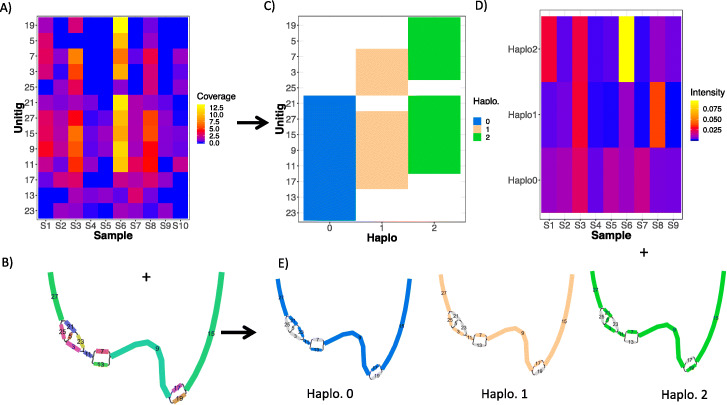
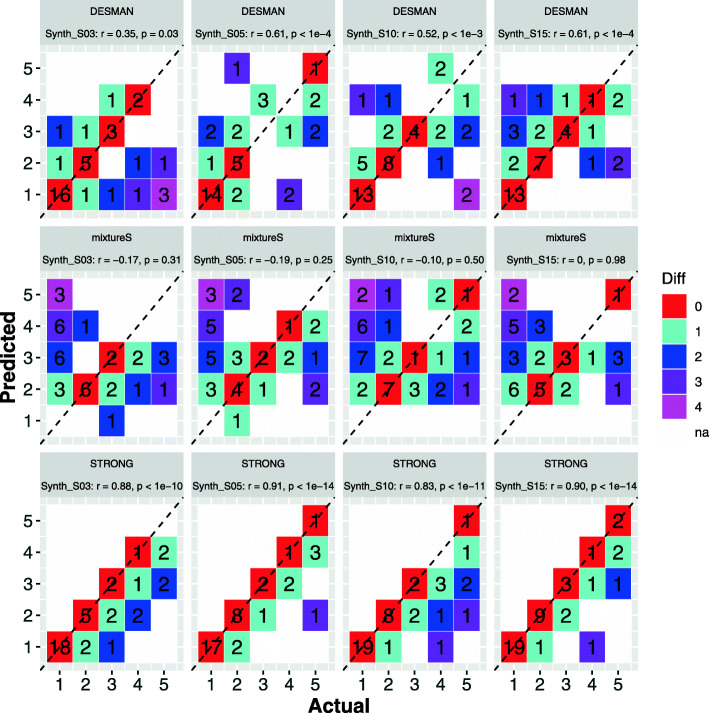
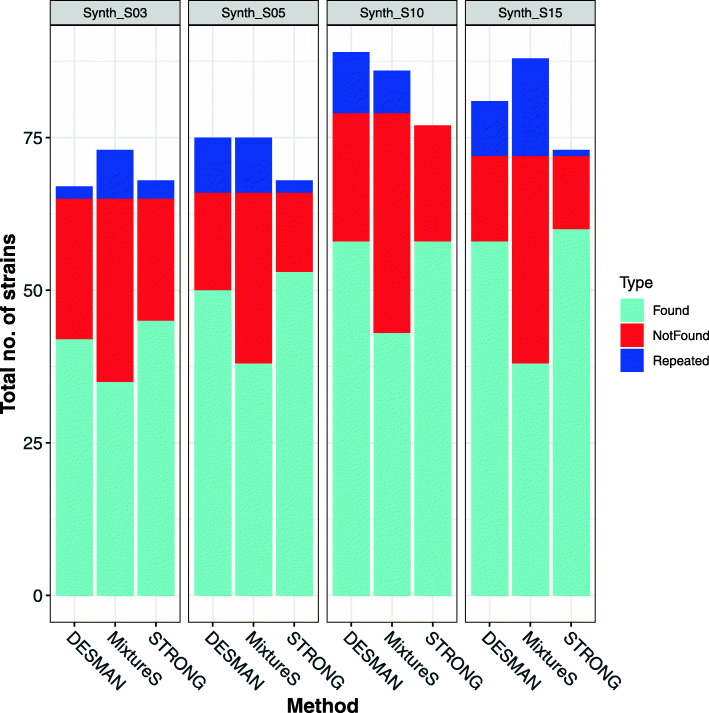
We introduce STrain Resolution ON assembly Graphs (STRONG), which identifies strains de novo, from multiple metagenome samples. STRONG performs coassembly, and binning into metagenome assembled genomes (MAGs), and stores the coassembly graph prior to variant simplification. This enables the subgraphs and their unitig per-sample coverages, for individual single-copy core genes (SCGs) in each MAG, to be extracted. A Bayesian algorithm, BayesPaths, determines the number of strains present, their haplotypes or sequences on the SCGs, and abundances. STRONG is validated using synthetic communities and for a real anaerobic digestor time series generates haplotypes that match those observed from long Nanopore reads.
Keywords: Assembly graph; Bayesian; Metagenome; Microbial community; Microbiome; Strains.
© 2021. The Author(s).
Conflict of interest statement
Aaron Darling is a cofounder of Longas Technologies Pty Ltd, a company that is developing synthetic long read sequencing technologies.
Figures
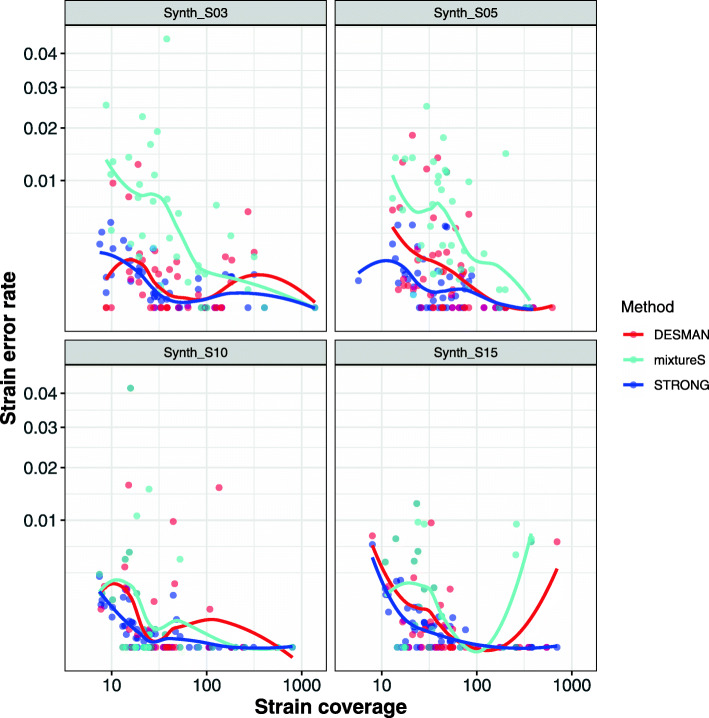
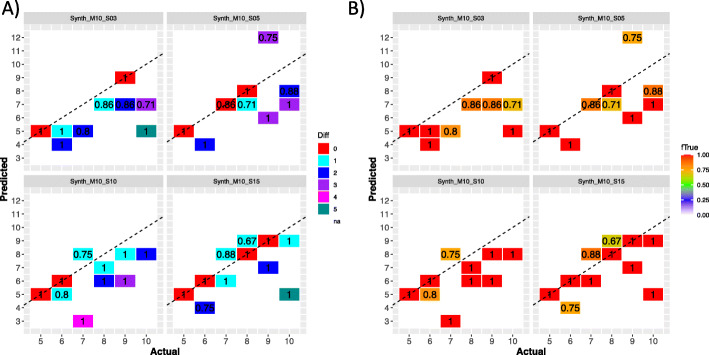
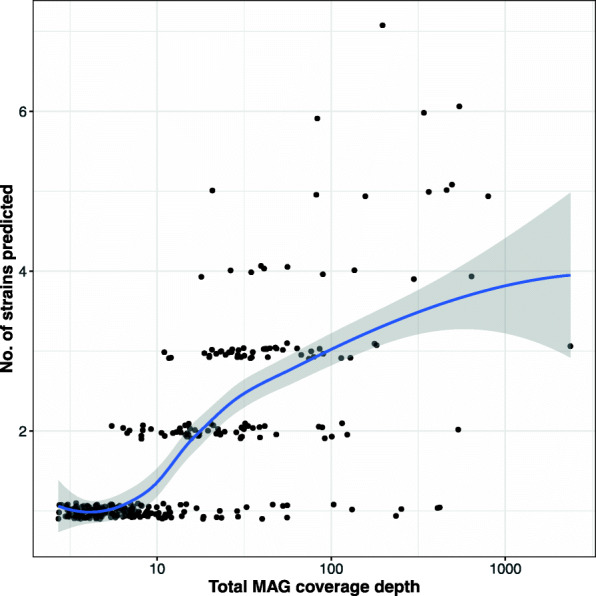
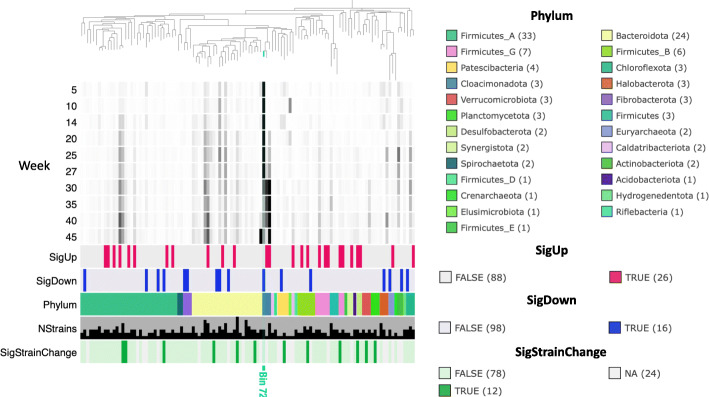
References
Publication types
MeSH terms
Grants and funding
- BBS/E/T/000PR9817/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MR/M50161X/1/MRC_/Medical Research Council/United Kingdom
- BB/N023285/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/L502029/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/K003240/2/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
