

Single-cell transcriptome of bronchoalveolar lavage fluid reveals sequential change of macrophages during SARS-CoV-2 infection in ferrets
- PMID: 34315893
- PMCID: PMC8316405
- DOI: 10.1038/s41467-021-24807-0
Single-cell transcriptome of bronchoalveolar lavage fluid reveals sequential change of macrophages during SARS-CoV-2 infection in ferrets
Abstract
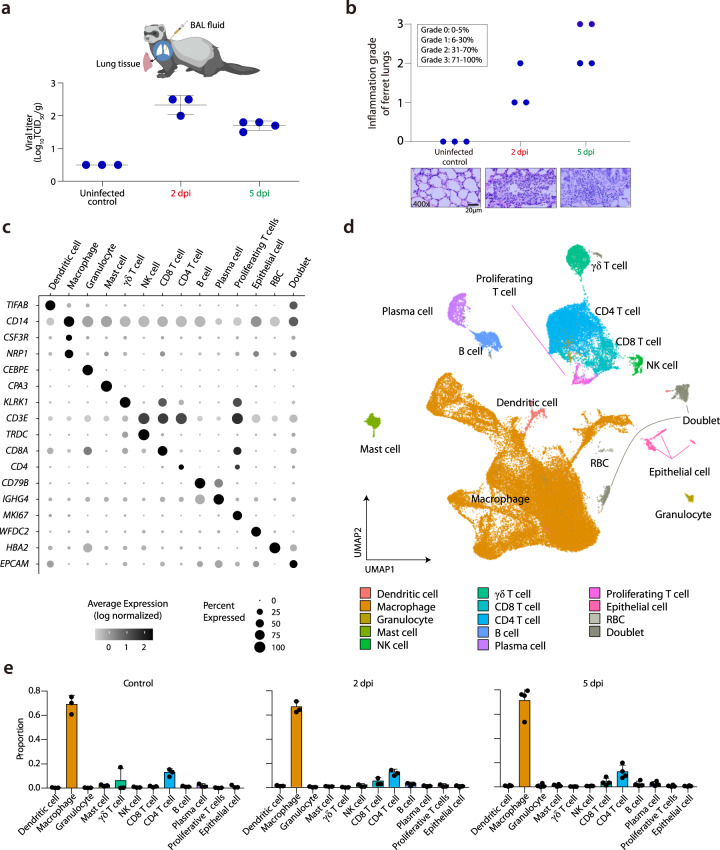
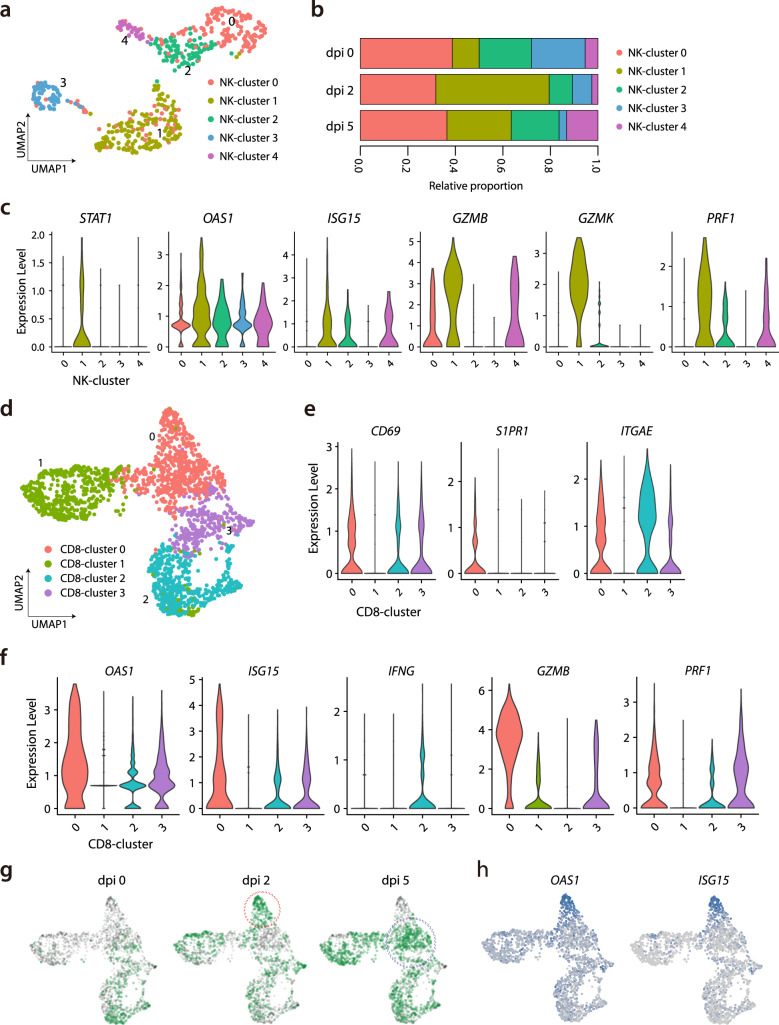
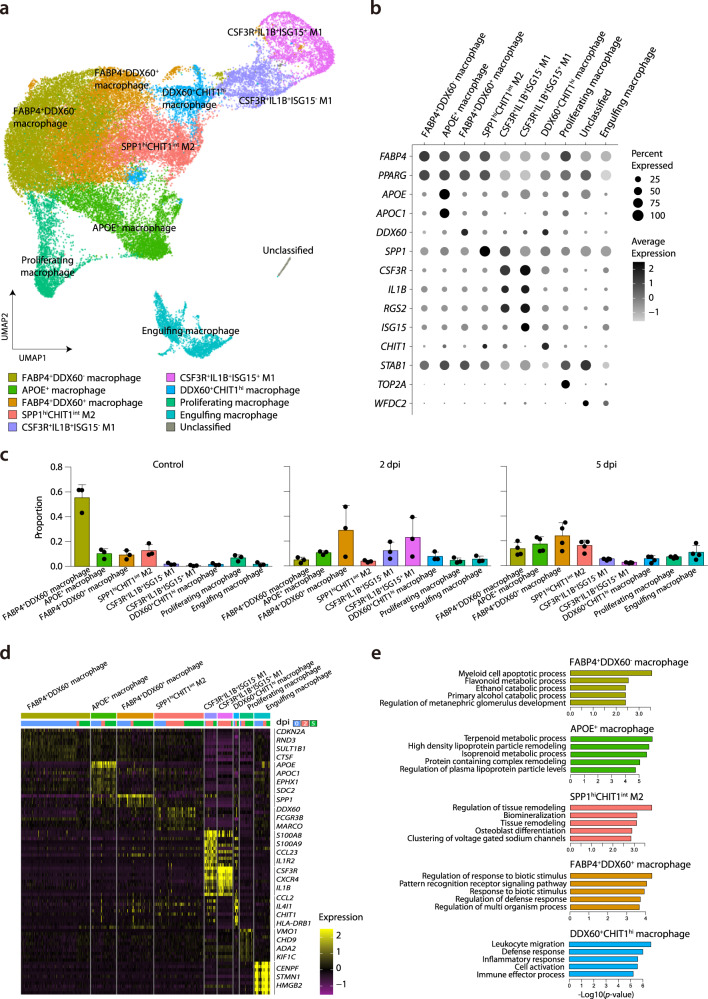
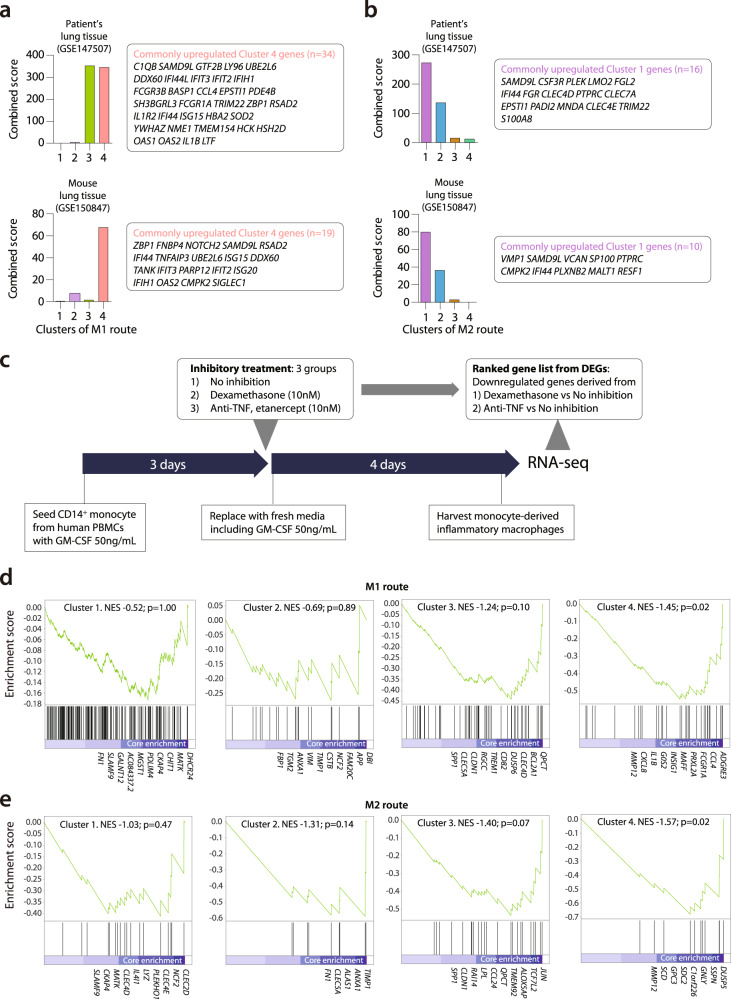
Few studies have used a longitudinal approach to describe the immune response to SARS-CoV-2 infection. Here, we perform single-cell RNA sequencing of bronchoalveolar lavage fluid cells longitudinally obtained from SARS-CoV-2-infected ferrets. Landscape analysis of the lung immune microenvironment shows distinct changes in cell proportions and characteristics compared to uninfected control, at 2 and 5 days post-infection (dpi). Macrophages are classified into 10 distinct subpopulations with transcriptome changes among monocyte-derived infiltrating macrophages and differentiated M1/M2 macrophages, notably at 2 dpi. Moreover, trajectory analysis reveals gene expression changes from monocyte-derived infiltrating macrophages toward M1 or M2 macrophages and identifies a macrophage subpopulation that has rapidly undergone SARS-CoV-2-mediated activation of inflammatory responses. Finally, we find that M1 or M2 macrophages show distinct patterns of gene modules downregulated by immune-modulatory drugs. Overall, these results elucidate fundamental aspects of the immune response dynamics provoked by SARS-CoV-2 infection.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
