

Functional and epigenetic phenotypes of humans and mice with DNMT3A Overgrowth Syndrome
- PMID: 34315901
- PMCID: PMC8316576
- DOI: 10.1038/s41467-021-24800-7
Functional and epigenetic phenotypes of humans and mice with DNMT3A Overgrowth Syndrome
Abstract
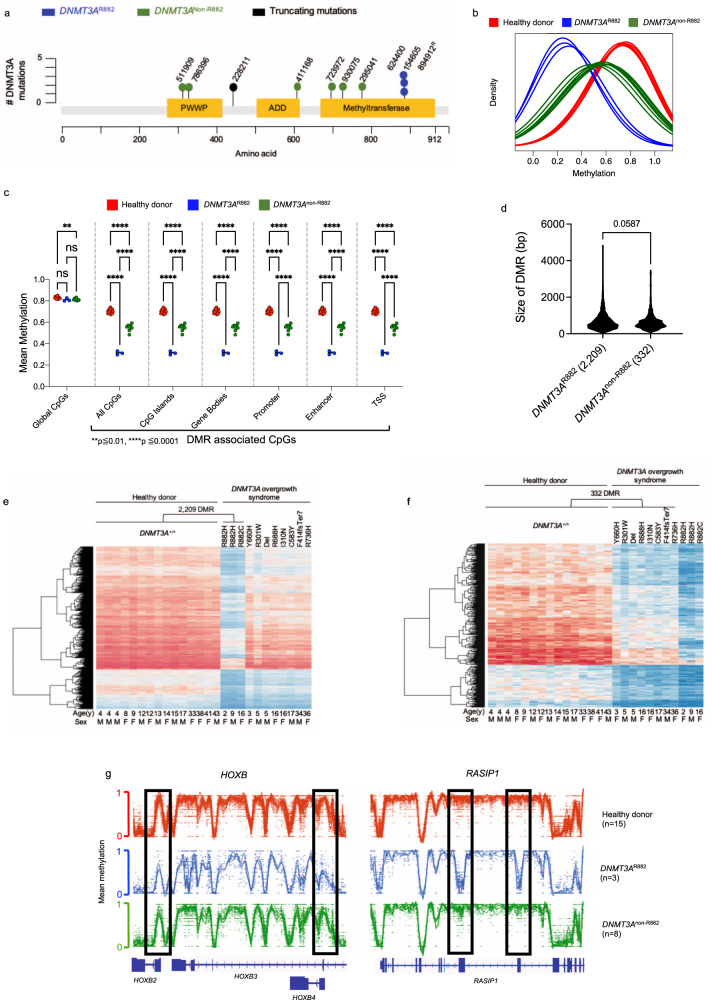
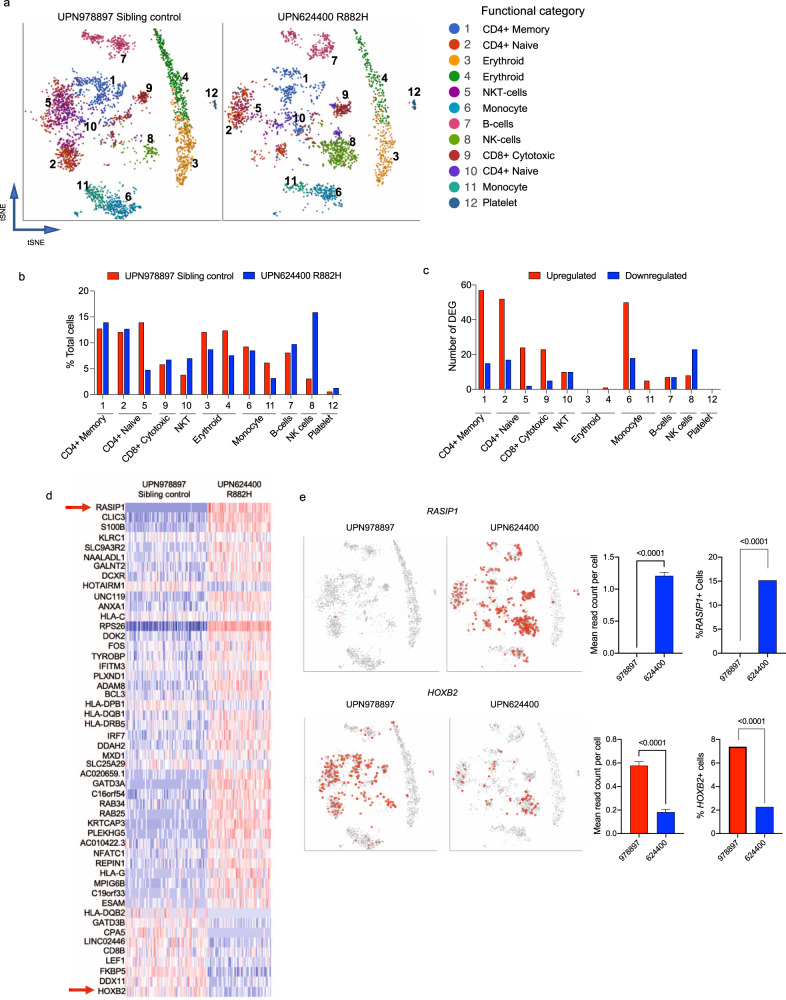
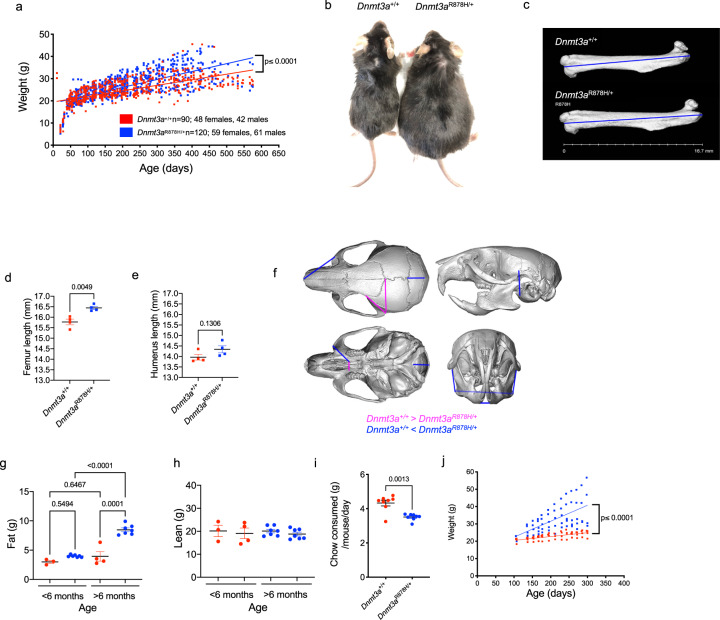
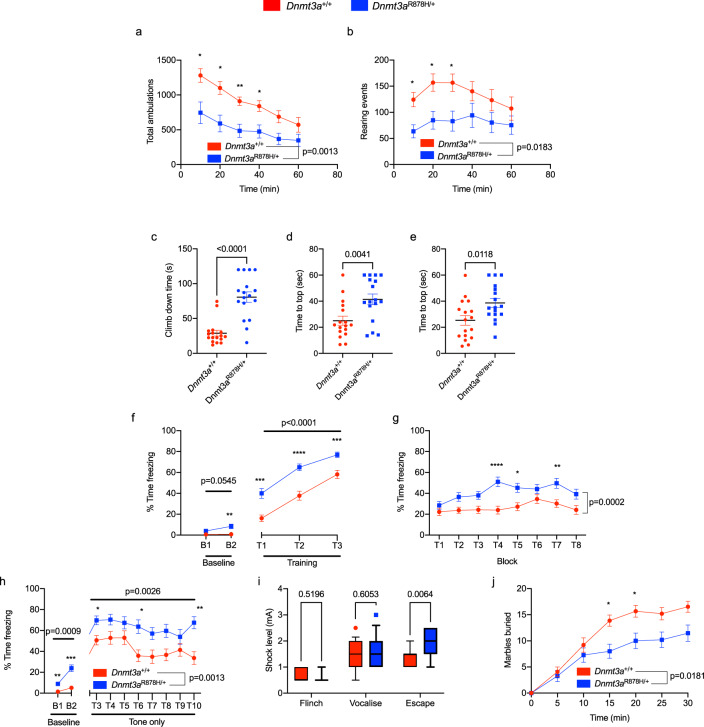
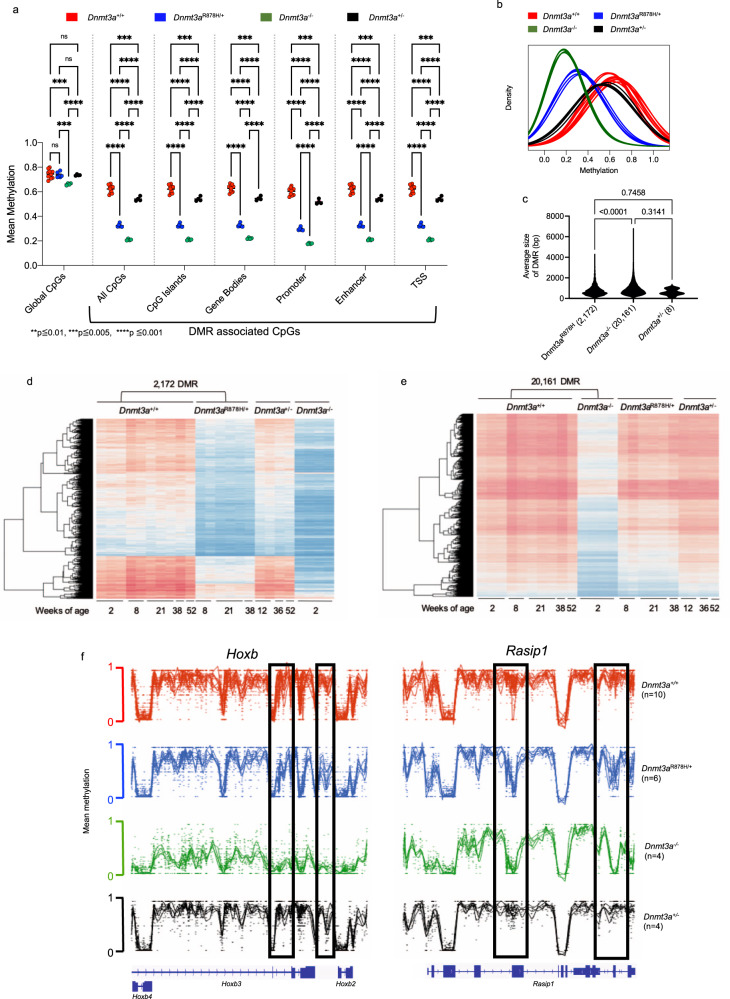
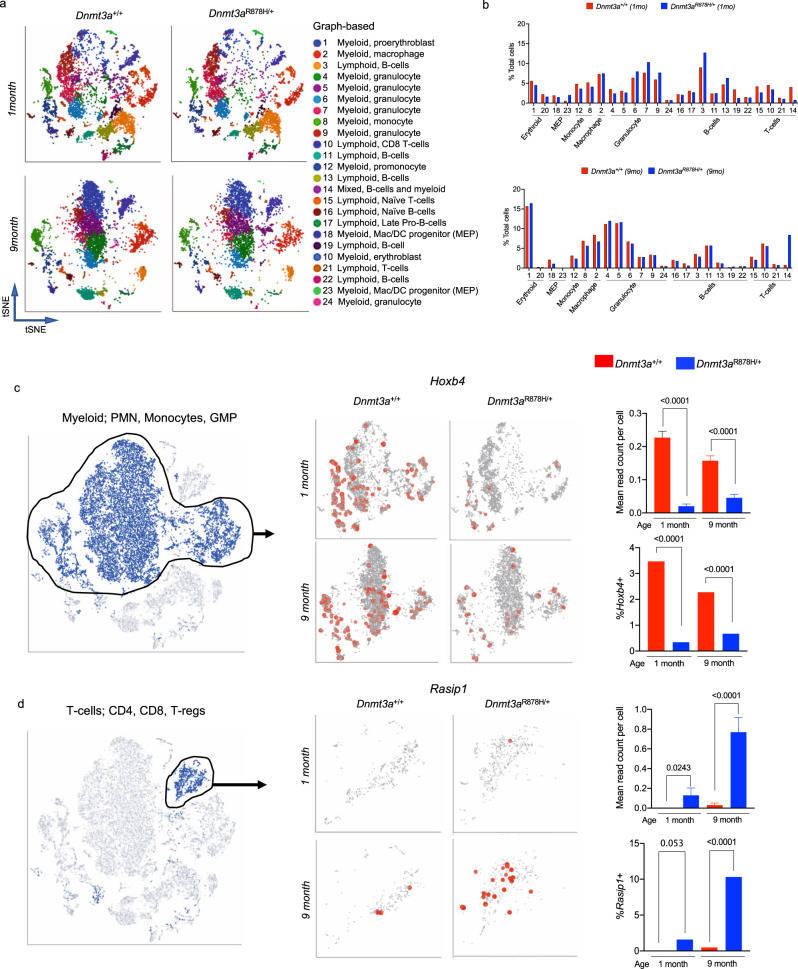
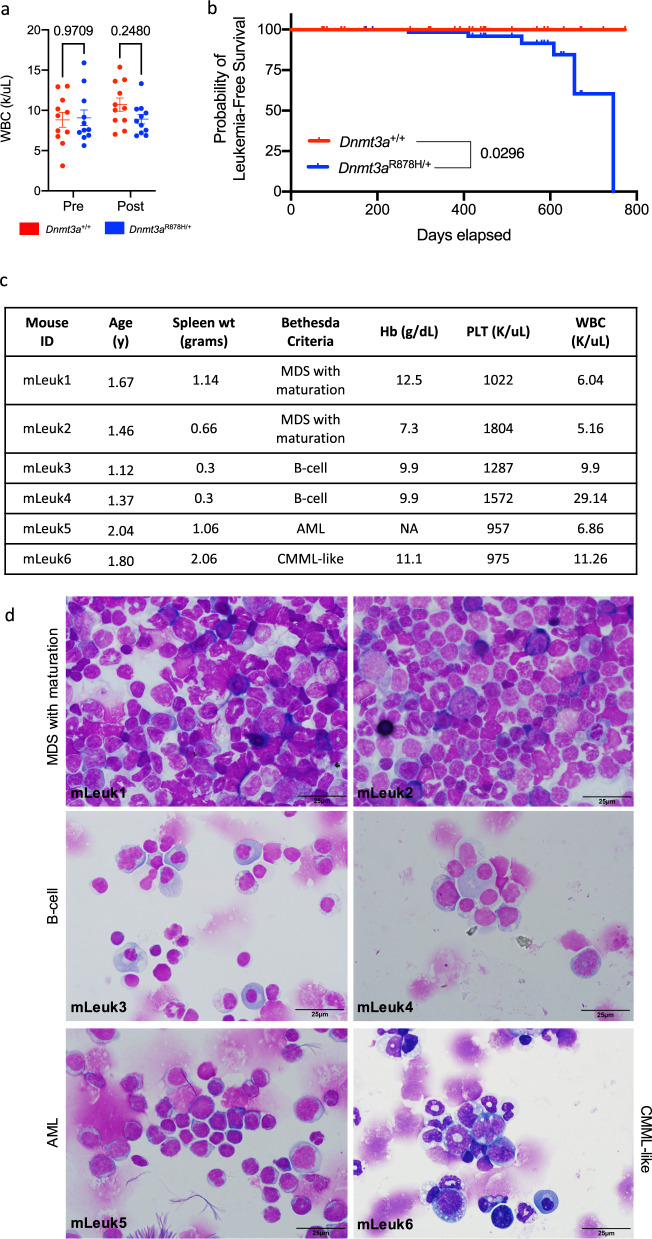
Germline pathogenic variants in DNMT3A were recently described in patients with overgrowth, obesity, behavioral, and learning difficulties (DNMT3A Overgrowth Syndrome/DOS). Somatic mutations in the DNMT3A gene are also the most common cause of clonal hematopoiesis, and can initiate acute myeloid leukemia (AML). Using whole genome bisulfite sequencing, we studied DNA methylation in peripheral blood cells of 11 DOS patients and found a focal, canonical hypomethylation phenotype, which is most severe with the dominant negative DNMT3AR882H mutation. A germline mouse model expressing the homologous Dnmt3aR878H mutation phenocopies most aspects of the human DOS syndrome, including the methylation phenotype and an increased incidence of spontaneous hematopoietic malignancies, suggesting that all aspects of this syndrome are caused by this mutation.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
