

Huntington disease: Advances in the understanding of its mechanisms
- PMID: 34316639
- PMCID: PMC8298812
- DOI: 10.1016/j.prdoa.2020.100056
Huntington disease: Advances in the understanding of its mechanisms
Abstract
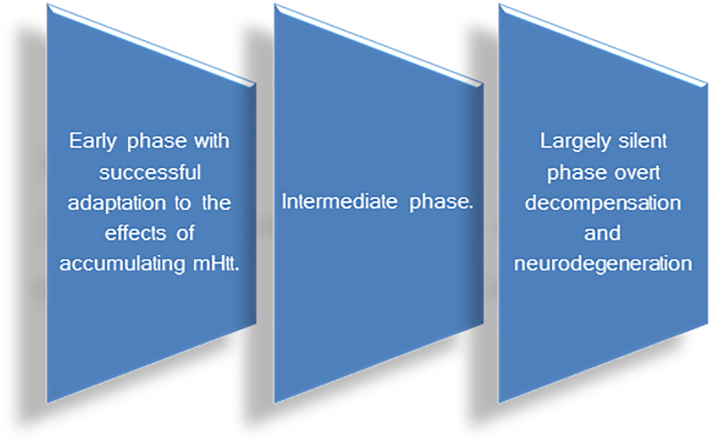
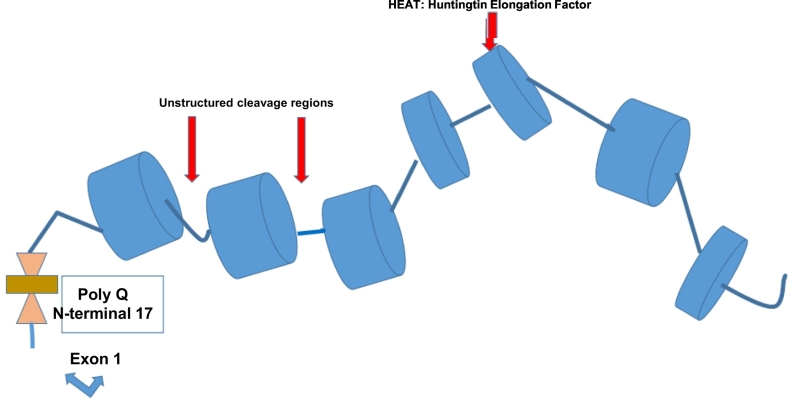
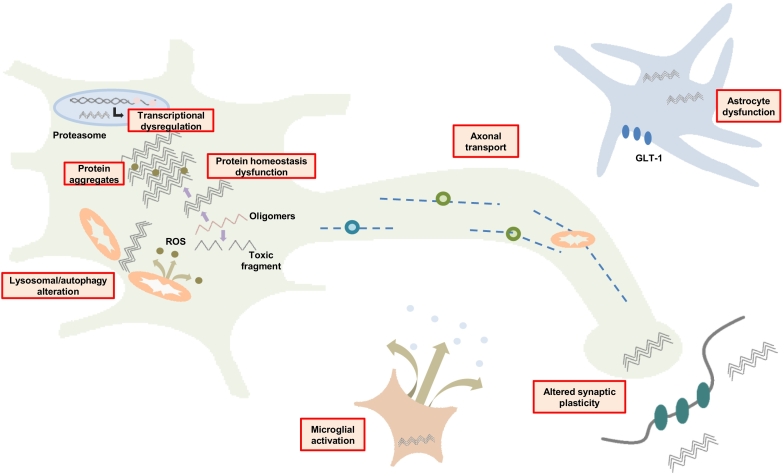
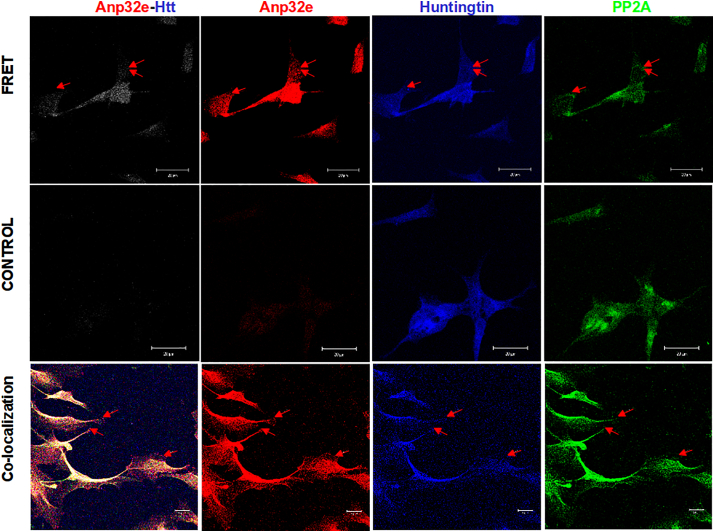
Huntington disease (HD) is a devastating monogenic autosomal dominant disorder. HD is caused by a CAG expansion in exon 1 of the gene coding for huntingtin, placed in the short arm of chromosome 4. Despite its well-defined genetic origin, the molecular and cellular mechanisms underlying the disease are unclear and complex. Here, we review some of the currently known functions of the wild-type huntingtin protein and discuss the deleterious effects that arise from the expansion of the CAG repeats, which are translated into an abnormally long polyglutamine tract. Also, we present a modern view on the molecular biology of HD as a representative of the group of polyglutamine diseases, with an emphasis on conformational changes of mutant huntingtin, disturbances in its cellular processing, and proteolytic stress in degenerating neurons. The main pathogenetic mechanisms of neurodegeneration in HD are discussed in detail, such as autophagy, impaired mitochondrial biogenesis, lysosomal dysfunction, organelle and protein transport, inflammation, oxidative stress, and transcription factor modulation. However, other unraveling mechanisms are still unknown. This practical and brief review summarizes some of the currently known functions of the wild-type huntingtin protein and the recent findings related to the mechanisms involved in HD pathogenesis.
Keywords: Huntington disease; Mechanisms; Pathophysiology.
© 2020 The Authors.
Figures
References
-
- Tabrizi S.J., Leavitt B.R., Landwehrmeyer G.B., Wild E.J., Saft C., Barker R.A., Blair N.F., Craufurd D., Priller J., Rickards H., Rosser A., Kordasiewicz H.B., Czech C., Swayze E.E., Norris D.A., Baumann T., Gerlach I., Schobel S.A., Paz E., Smith A.V., Bennett C.F., Lane R.M. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 2019;380:2307–2316. doi: 10.1056/NEJMoa1900907. - DOI - PubMed
-
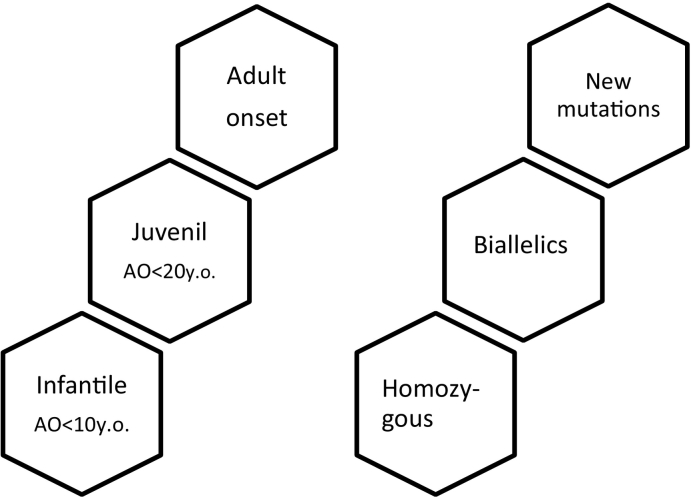
- Quarrell O.W.J., Nance M.A., Nopoulos P., Reilmann R., Oosterloo M., Tabrizi S.J., Furby H., Saft C., Roos R.A.C., Squitieri F., Landwehrmeyer G.B., Burgunder J.M. Juvenile Huntington Disease Working Group of the European Huntington Disease Network. Defining pediatric Huntington disease: time to abandon the term juvenile Huntington disease? Mov. Disord. 2019;34:584–585. doi: 10.1002/mds.27640. - DOI - PubMed
-
- Fusilli C., Migliore S., Mazza T., Consoli F., De Luca A., Barbagallo G., Ciammola A., Gatto E.M., Cesarini M., Etcheverry J.L., Parisi V., Al-Oraimi M., Al-Harrasi S., Al-Salmi Q., Marano M., Vonsattel J.G., Sabatini U., Landwehrmeyer G.B., Squitieri F. Biological and clinical manifestations of juvenile Huntington’s disease: a retrospective analysis. Lancet Neurol. 2018;17:986–993. doi: 10.1016/S1474-4422(18)30294-1. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
