

Improving Measures of Chemical Structural Similarity Using Machine Learning on Chemical-Genetic Interactions
- PMID: 34318674
- PMCID: PMC8479812
- DOI: 10.1021/acs.jcim.0c00993
Improving Measures of Chemical Structural Similarity Using Machine Learning on Chemical-Genetic Interactions
Abstract
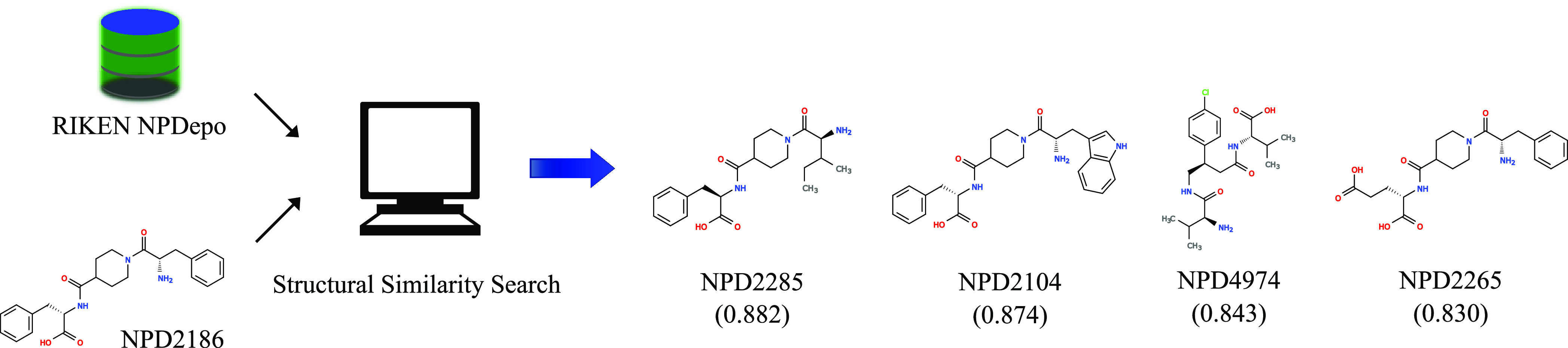
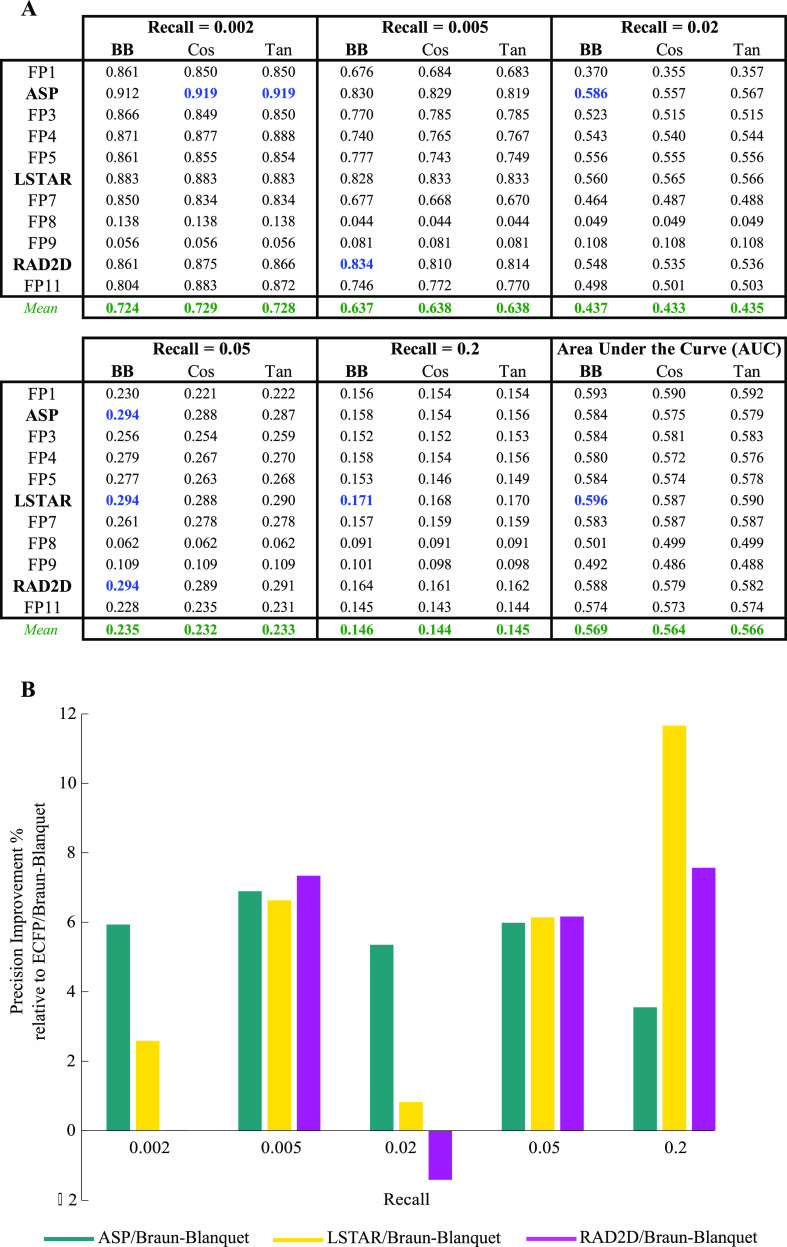
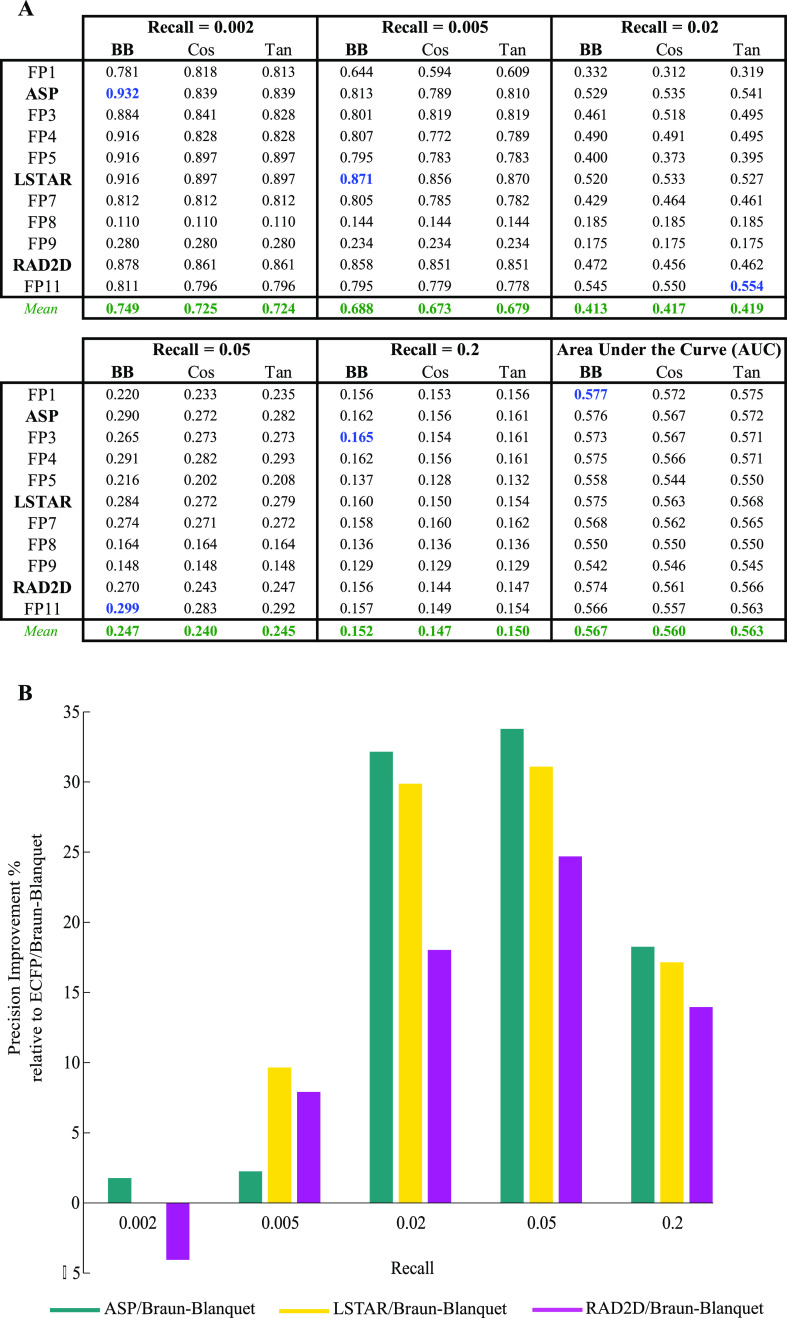
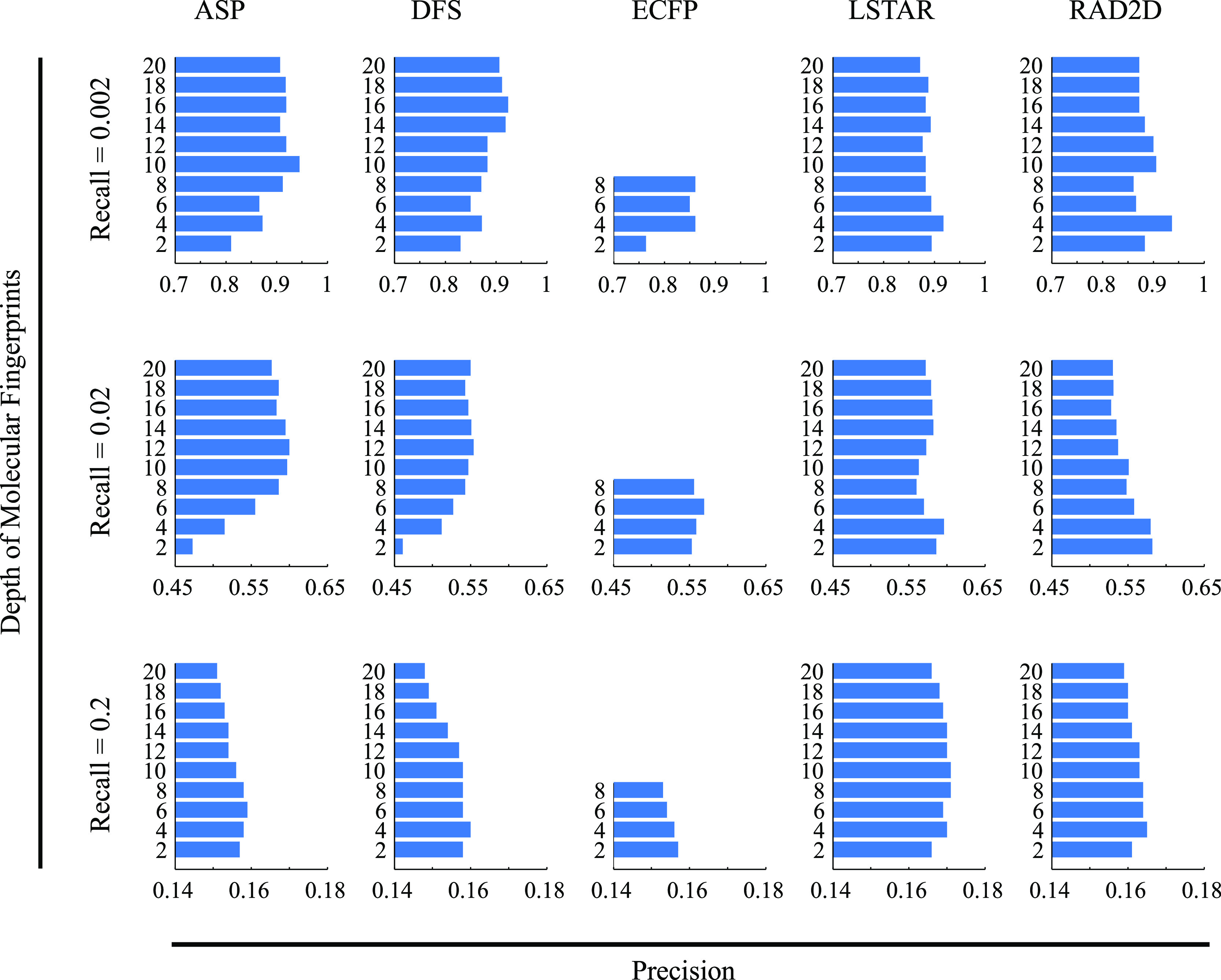
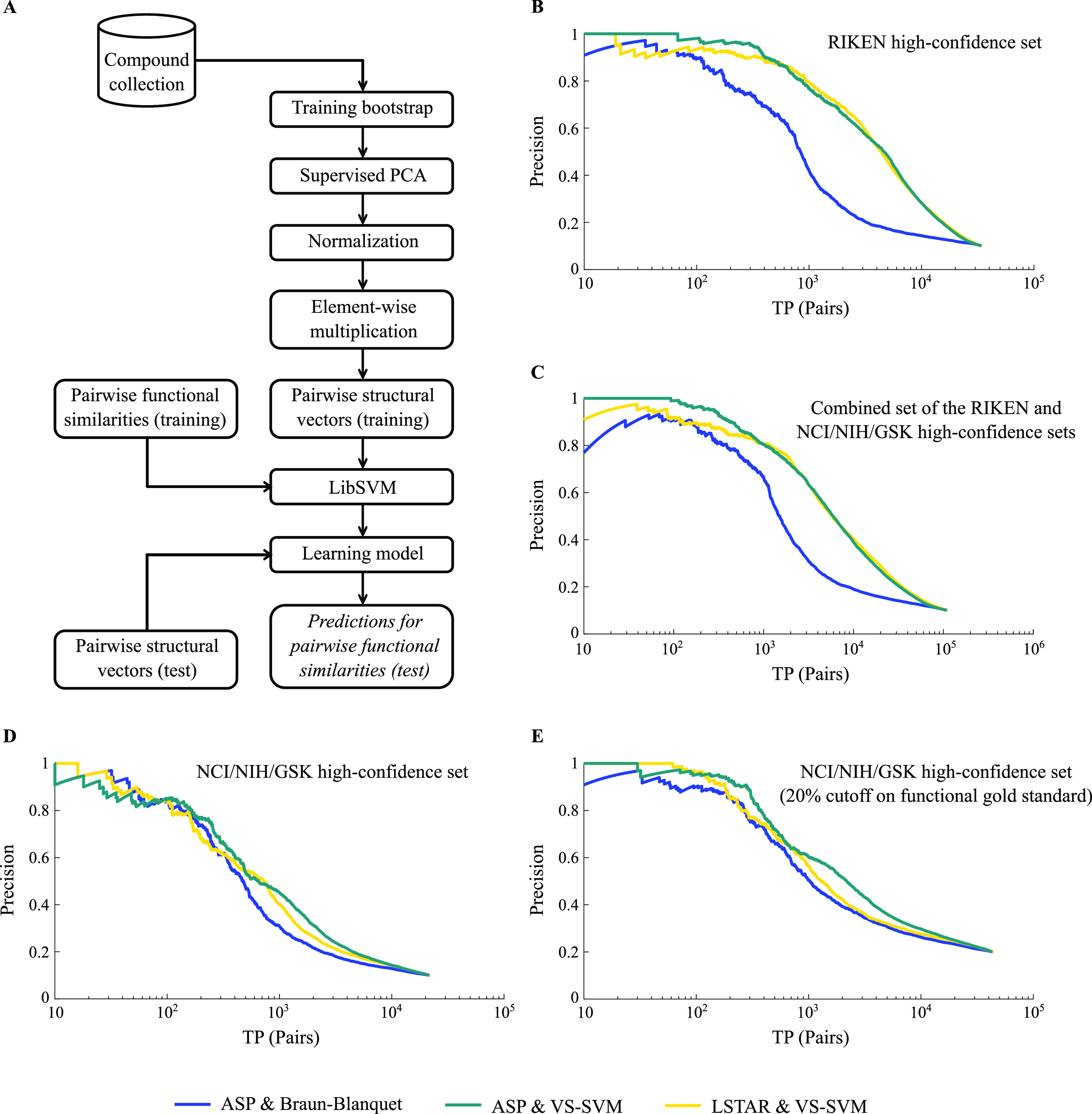
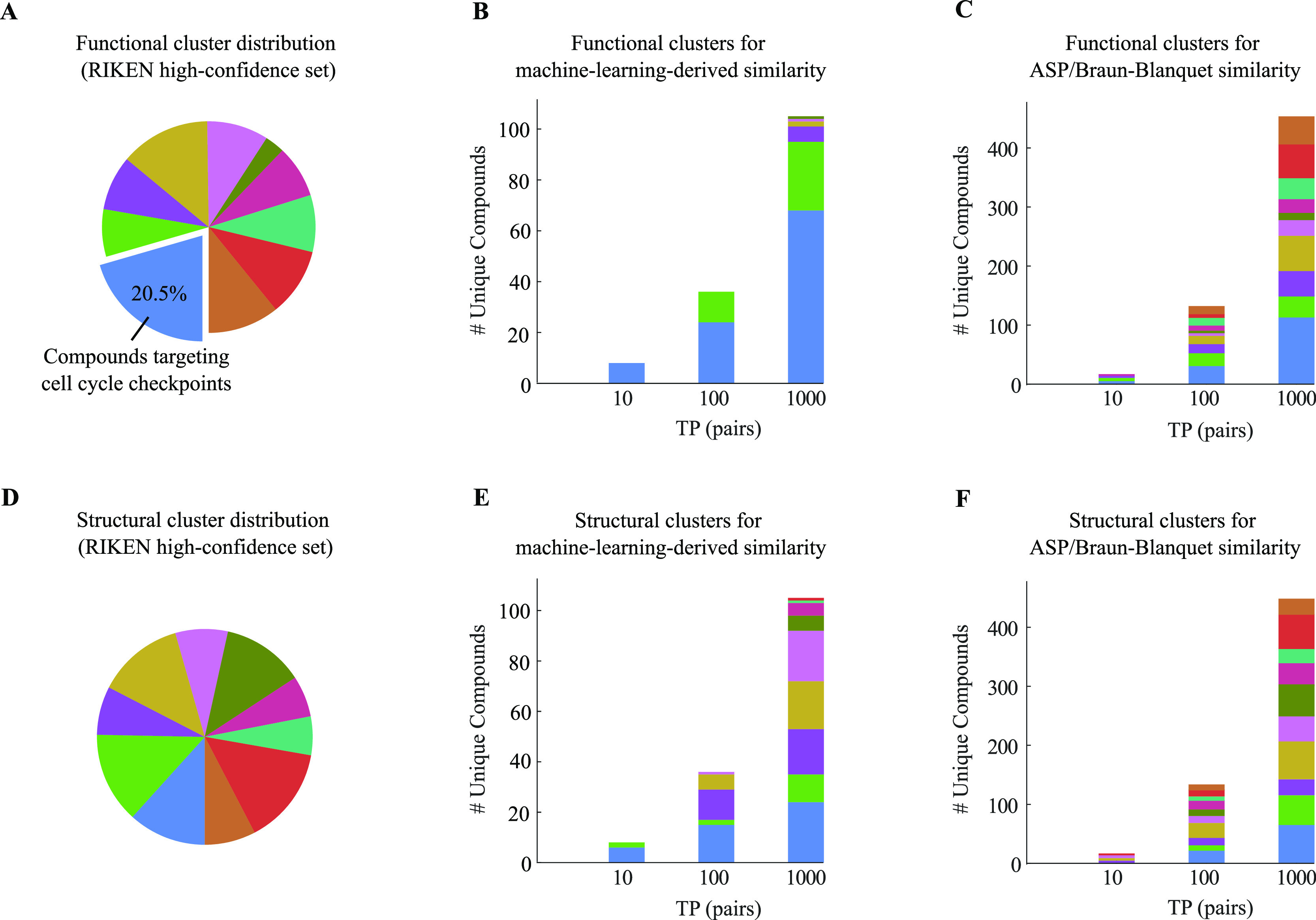
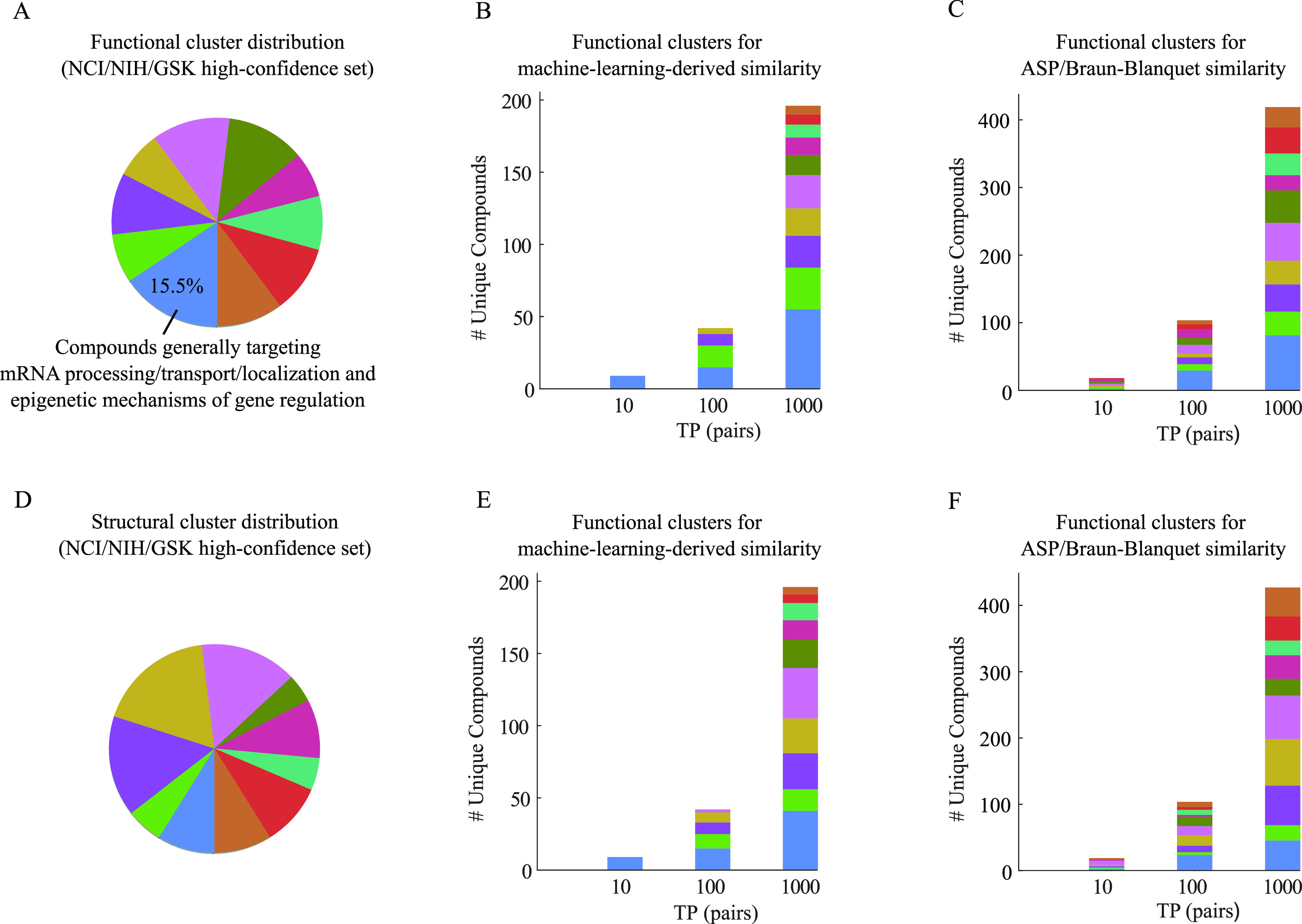
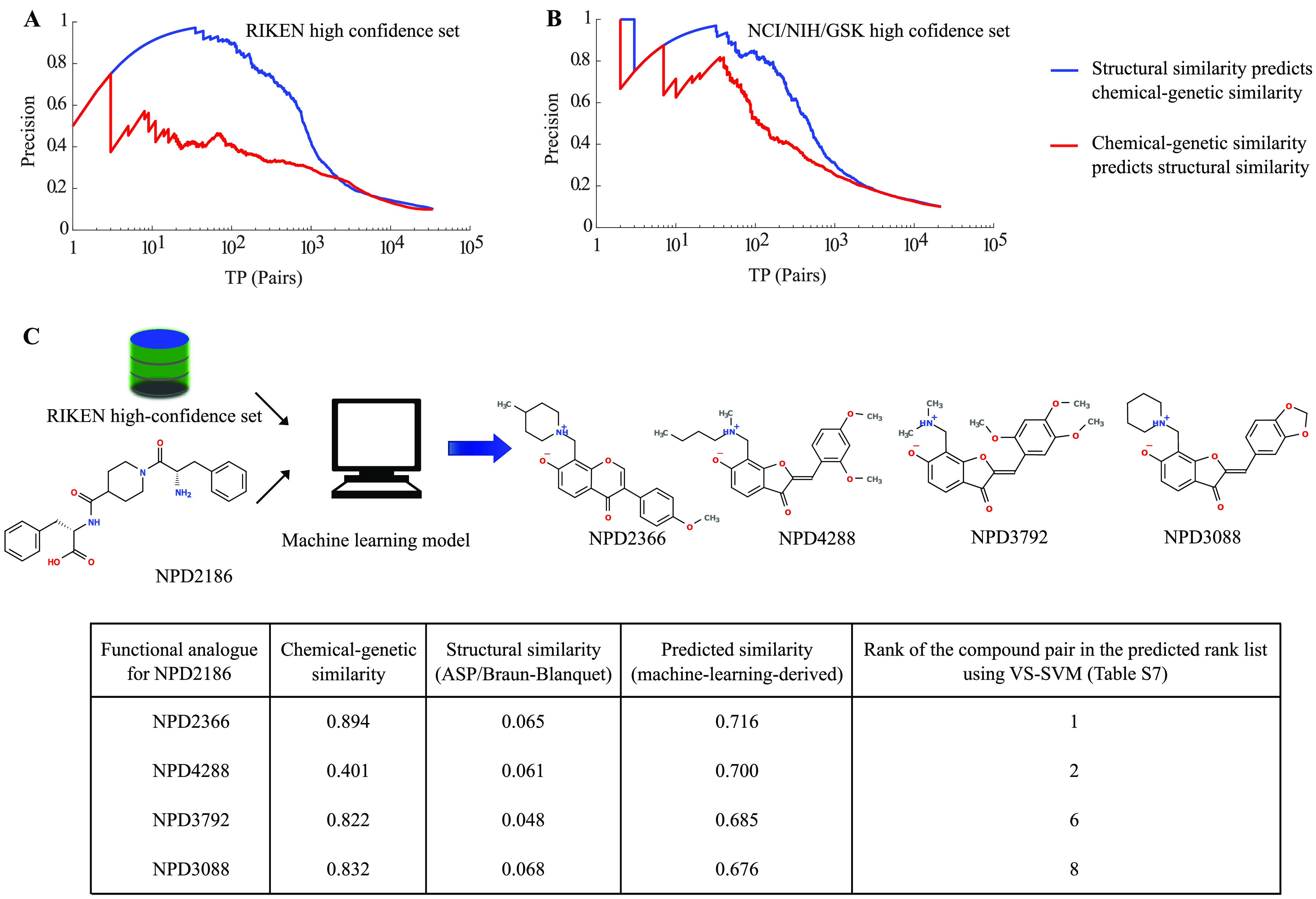
A common strategy for identifying molecules likely to possess a desired biological activity is to search large databases of compounds for high structural similarity to a query molecule that demonstrates this activity, under the assumption that structural similarity is predictive of similar biological activity. However, efforts to systematically benchmark the diverse array of available molecular fingerprints and similarity coefficients have been limited by a lack of large-scale datasets that reflect biological similarities of compounds. To elucidate the relative performance of these alternatives, we systematically benchmarked 11 different molecular fingerprint encodings, each combined with 13 different similarity coefficients, using a large set of chemical-genetic interaction data from the yeast Saccharomyces cerevisiae as a systematic proxy for biological activity. We found that the performance of different molecular fingerprints and similarity coefficients varied substantially and that the all-shortest path fingerprints paired with the Braun-Blanquet similarity coefficient provided superior performance that was robust across several compound collections. We further proposed a machine learning pipeline based on support vector machines that offered a fivefold improvement relative to the best unsupervised approach. Our results generally suggest that using high-dimensional chemical-genetic data as a basis for refining molecular fingerprints can be a powerful approach for improving prediction of biological functions from chemical structures.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Johnson M. A.; Maggiora G. M.. Concepts and Applications of Molecular Similarity; Wiley, 1990. (accessed July 17, 2020). https://agris.fao.org/agris-search/search.do?recordID=US201300674768.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
