

Ebola Virus Requires Phosphatidylserine Scrambling Activity for Efficient Budding and Optimal Infectivity
- PMID: 34319156
- PMCID: PMC8475530
- DOI: 10.1128/JVI.01165-21
Ebola Virus Requires Phosphatidylserine Scrambling Activity for Efficient Budding and Optimal Infectivity
Abstract
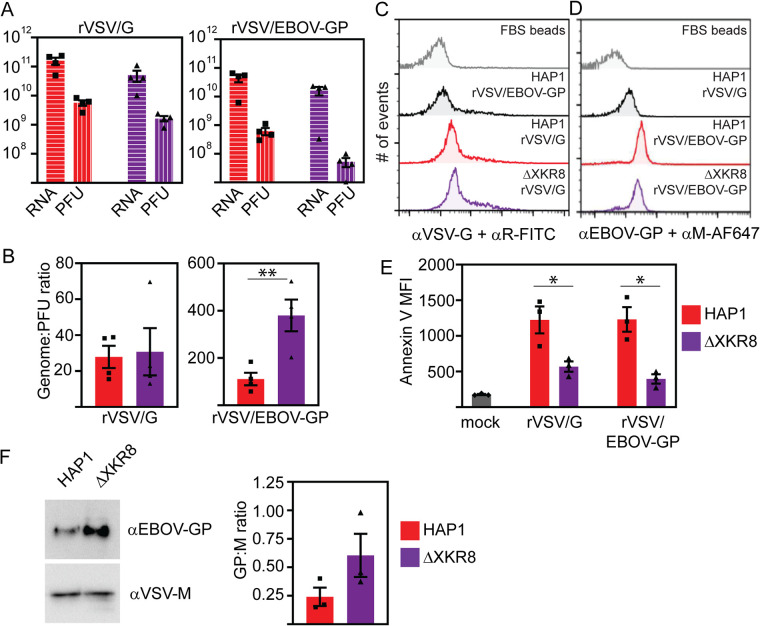
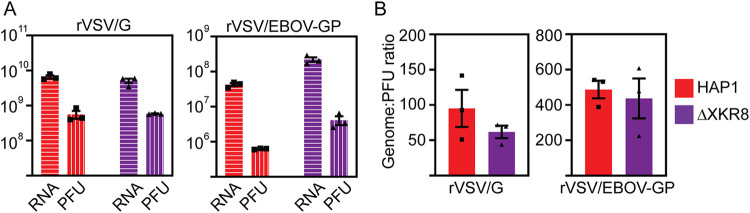
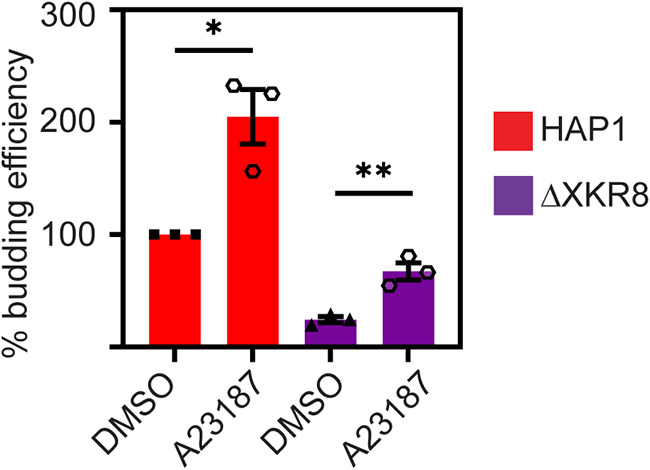
Ebola virus (EBOV) attaches to target cells using two categories of cell surface receptors: C-type lectins and phosphatidylserine (PS) receptors. PS receptors typically bind to apoptotic cell membrane PS and orchestrate the uptake and clearance of apoptotic debris. Many enveloped viruses also contain exposed PS and can therefore exploit these receptors for cell entry. Viral infection can induce PS externalization in host cells, resulting in increased outer PS levels on budding virions. Scramblase enzymes carry out cellular PS externalization; thus, we targeted these proteins in order to manipulate viral envelope PS levels. We investigated two scramblases previously identified to be involved in EBOV PS levels, transmembrane protein 16F and Xk-related protein 8 (XKR8), as possible mediators of cellular and viral envelope surface PS levels during the replication of recombinant vesicular stomatitis virus containing its native glycoprotein (rVSV/G) or the EBOV glycoprotein (rVSV/EBOV-GP). We found that rVSV/G and rVSV/EBOV-GP virions produced in XKR8 knockout cells contain decreased levels of PS on their surfaces, and the PS-deficient rVSV/EBOV-GP virions are 70% less efficient at infecting cells through PS receptors. We also observed reduced rVSV and EBOV virus-like particle (VLP) budding in ΔXKR8 cells. Deletion of XKR8 in HAP1 cells reduced rVSV/G and rVSV/EBOV-GP budding by 60 and 65%, respectively, and reduced Ebola VLP budding more than 60%. We further demonstrated that caspase cleavage of XKR8 is required to promote budding. This suggests that XKR8, in addition to mediating virion PS levels, may also be critical for enveloped virus budding at the plasma membrane. IMPORTANCE Within the last decade, countries in western and central Africa have experienced the most widespread and deadly Ebola outbreaks since Ebola virus was identified in 1976. While outbreaks are primarily attributed to zoonotic transfer events, new evidence is emerging outbreaks may be caused by a combination of spillover events and viral latency or persistence in survivors. The possibility that Ebola virus can remain dormant and then reemerge in survivors highlights the critical need to prevent the virus from entering and establishing infection in human cells. Thus far, host cell scramblases TMEM16F and XKR8 have been implicated in Ebola envelope surface phosphatidylserine (PS) and cell entry using PS receptors. We assessed the contributions of these proteins using CRISPR knockout cells and two EBOV models: rVSV/EBOV-GP and EBOV VLPs. We observed that XKR8 is required for optimal EBOV envelope PS levels and infectivity and particle budding across all viral models.
Keywords: Ebola; XKR8; budding; phosphatidylserine.
Figures

Similar articles
-
Ebola virus requires a host scramblase for externalization of phosphatidylserine on the surface of viral particles.PLoS Pathog. 2018 Jan 16;14(1):e1006848. doi: 10.1371/journal.ppat.1006848. eCollection 2018 Jan. PLoS Pathog. 2018. PMID: 29338048 Free PMC article.
-
Molecular Mechanism of Externalization of Phosphatidylserine on the Surface of Ebola Virus Particles.DNA Cell Biol. 2019 Feb;38(2):115-120. doi: 10.1089/dna.2018.4485. Epub 2019 Jan 7. DNA Cell Biol. 2019. PMID: 30615471 Review.
-
TIM-1 serves as a receptor for Ebola virus in vivo, enhancing viremia and pathogenesis.PLoS Negl Trop Dis. 2019 Jun 26;13(6):e0006983. doi: 10.1371/journal.pntd.0006983. eCollection 2019 Jun. PLoS Negl Trop Dis. 2019. PMID: 31242184 Free PMC article.
-
Host Cell Plasma Membrane Phosphatidylserine Regulates the Assembly and Budding of Ebola Virus.J Virol. 2015 Sep;89(18):9440-53. doi: 10.1128/JVI.01087-15. Epub 2015 Jul 1. J Virol. 2015. PMID: 26136573 Free PMC article.
-
[Research progress on ebola virus glycoprotein].Bing Du Xue Bao. 2013 Mar;29(2):233-7. Bing Du Xue Bao. 2013. PMID: 23757858 Review. Chinese.
Cited by
-
The Art of Viral Membrane Fusion and Penetration.Subcell Biochem. 2023;106:113-152. doi: 10.1007/978-3-031-40086-5_4. Subcell Biochem. 2023. PMID: 38159225
-
HAP1, a new revolutionary cell model for gene editing using CRISPR-Cas9.Front Cell Dev Biol. 2023 Mar 3;11:1111488. doi: 10.3389/fcell.2023.1111488. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 36936678 Free PMC article. Review.
-
The use of viral vectors in vaccine development.NPJ Vaccines. 2022 Jul 4;7(1):75. doi: 10.1038/s41541-022-00503-y. NPJ Vaccines. 2022. PMID: 35787629 Free PMC article. Review.
-
Enveloped RNA virus utilization of phosphatidylserine receptors: Advantages of exploiting a conserved, widely available mechanism of entry.PLoS Pathog. 2021 Sep 23;17(9):e1009899. doi: 10.1371/journal.ppat.1009899. eCollection 2021 Sep. PLoS Pathog. 2021. PMID: 34555126 Free PMC article. Review. No abstract available.
-
The Effects of Viral Structural Proteins on Acidic Phospholipids in Host Membranes.Viruses. 2024 Oct 31;16(11):1714. doi: 10.3390/v16111714. Viruses. 2024. PMID: 39599829 Free PMC article. Review.
References
-
- CDC. 2018. Ebola virus disease distribution map: cases of Ebola virus disease in Africa since 1976. https://www.cdc.gov/vhf/ebola/history/distribution-map.html. Accessed 23 August 2018.
-
- Dokubo EK, Wendland A, Mate SE, Ladner JT, Hamblion EL, Raftery P, Blackley DJ, Laney AS, Mahmoud N, Wayne-Davies G, Hensley L, Stavale E, Fakoli L, Gregory C, Chen TH, Koryon A, Roth Allen D, Mann J, Hickey A, Saindon J, Badini M, Baller A, Clement P, Bolay F, Wapoe Y, Wiley MR, Logue J, Dighero-Kemp B, Higgs E, Gasasira A, Williams DE, Dahn B, Kateh F, Nyenswah T, Palacios G, Fallah MP. 2018. Persistence of Ebola virus after the end of widespread transmission in Liberia: an outbreak report. Lancet Infect Dis 18:1015–1024. 10.1016/S1473-3099(18)30417-1. - DOI - PubMed
-
- WHO. 2018. Ebola virus disease. http://www.who.int/news-room/fact-sheets/detail/ebola-virus-disease. Accessed 23 August 2018.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
