

Yersinia pseudotuberculosis YopJ Limits Macrophage Response by Downregulating COX-2-Mediated Biosynthesis of PGE2 in a MAPK/ERK-Dependent Manner
- PMID: 34319170
- PMCID: PMC8552654
- DOI: 10.1128/Spectrum.00496-21
Yersinia pseudotuberculosis YopJ Limits Macrophage Response by Downregulating COX-2-Mediated Biosynthesis of PGE2 in a MAPK/ERK-Dependent Manner
Erratum in
-
Erratum for Sheppe et al., "Yersinia pseudotuberculosis YopJ Limits Macrophage Response by Downregulating COX-2-Mediated Biosynthesis of PGE2 in a MAPK/ERK-Dependent Manner".Microbiol Spectr. 2023 Jun 15;11(3):e0399722. doi: 10.1128/spectrum.03997-22. Epub 2023 Apr 6. Microbiol Spectr. 2023. PMID: 37022198 Free PMC article. No abstract available.
Abstract
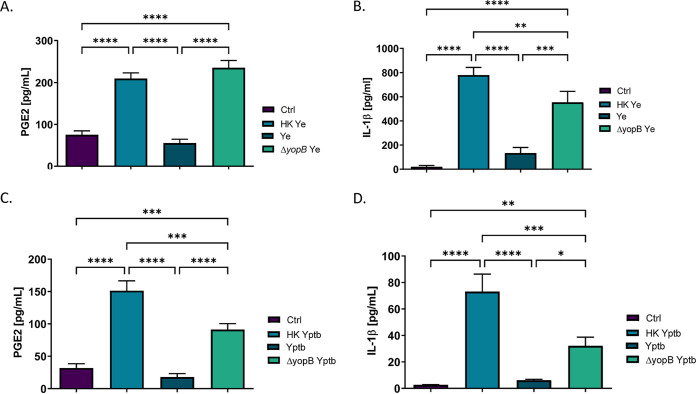
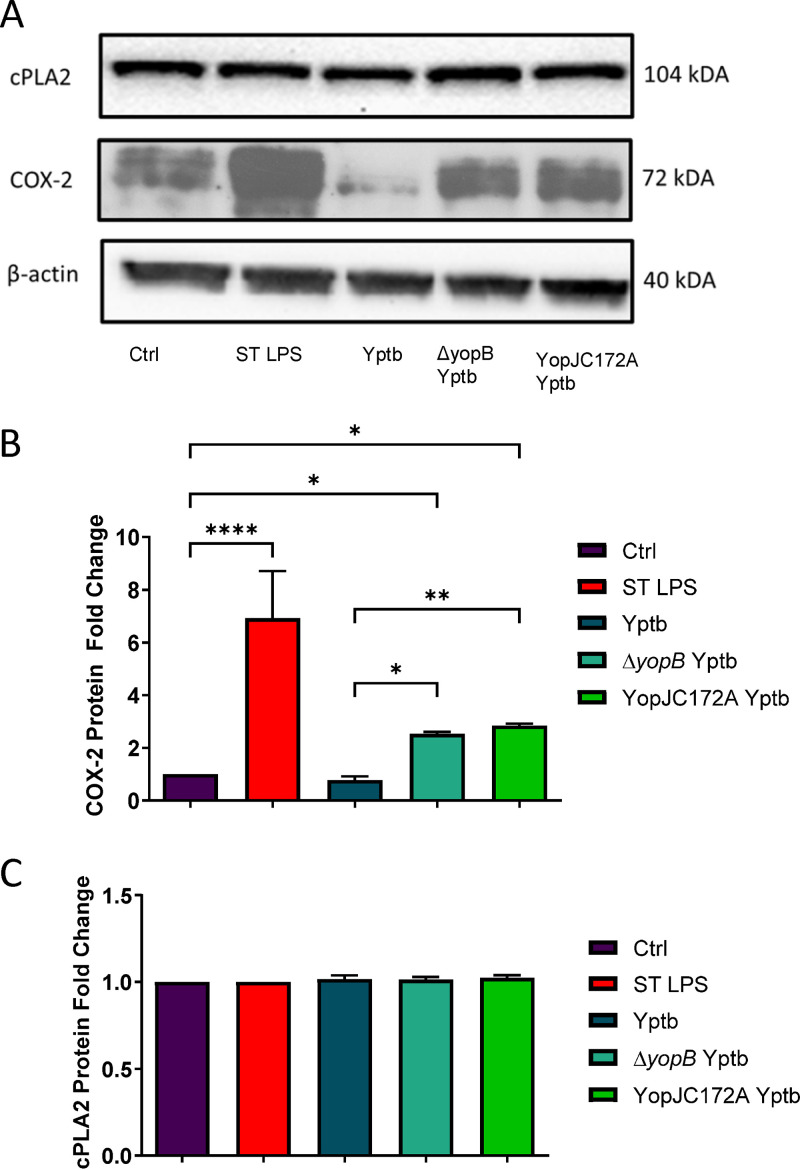
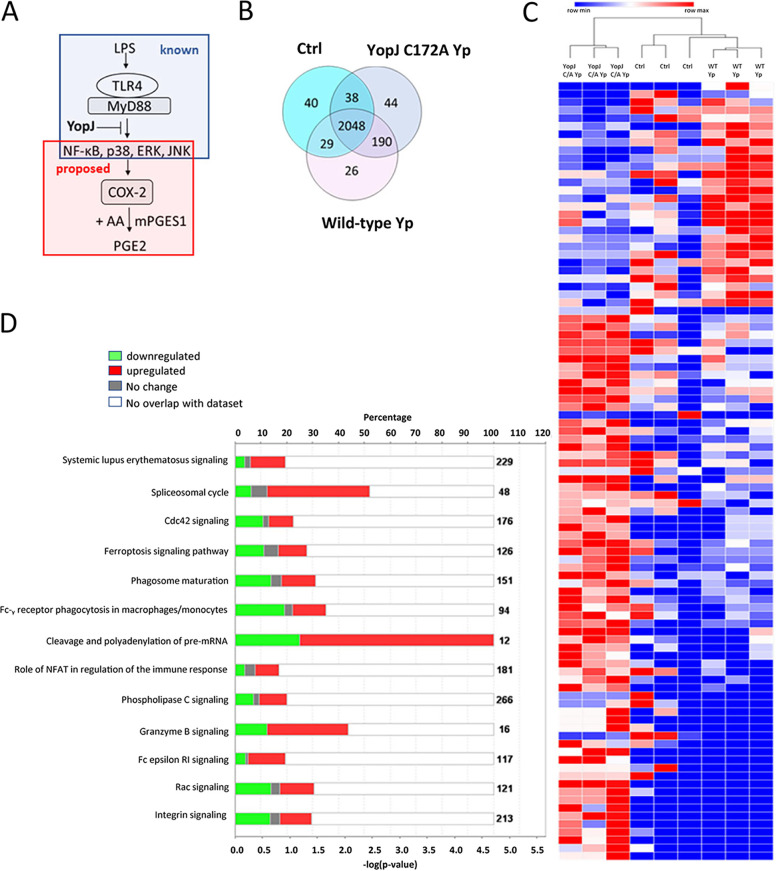
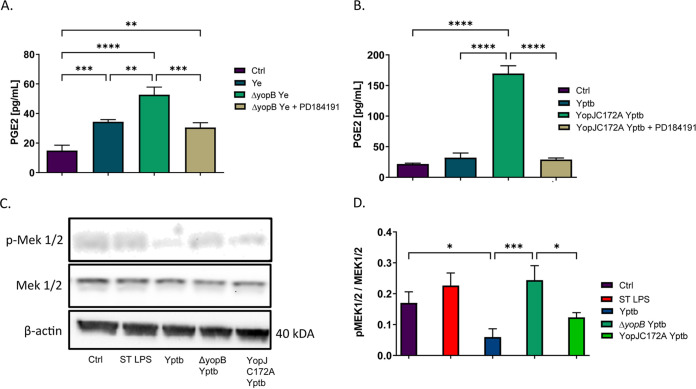
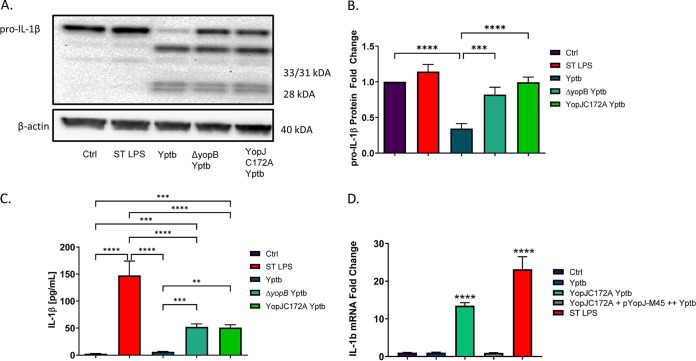
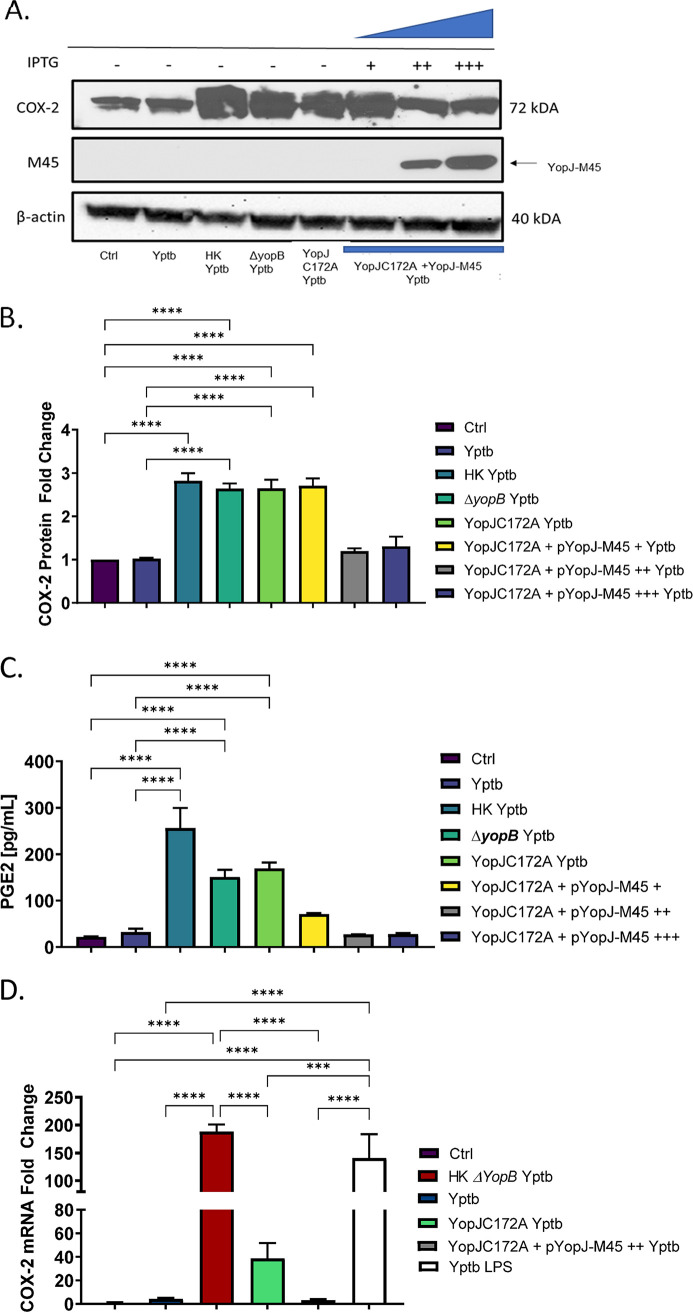
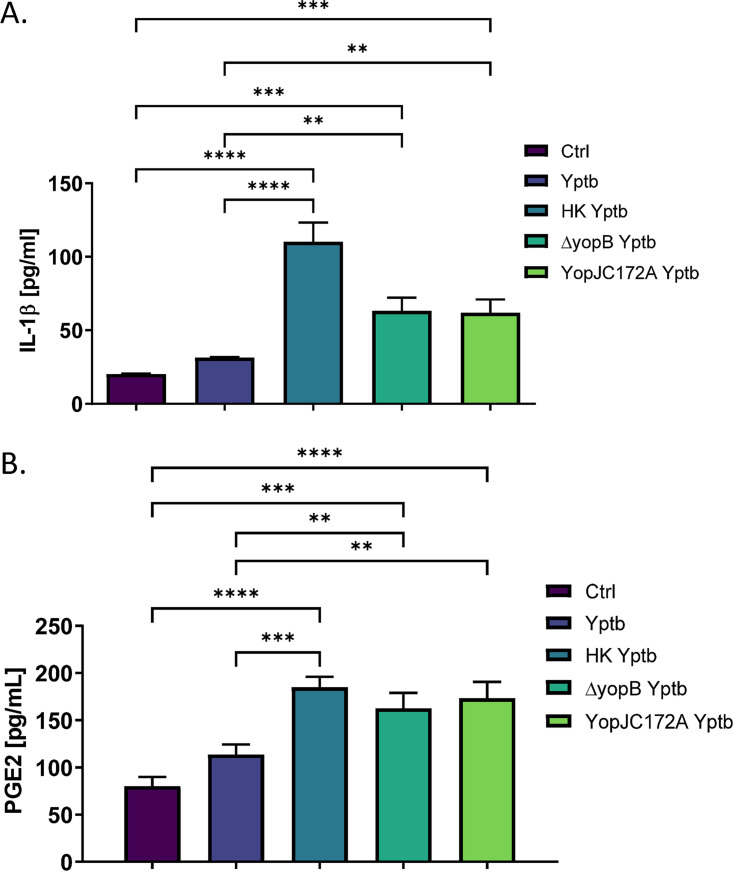
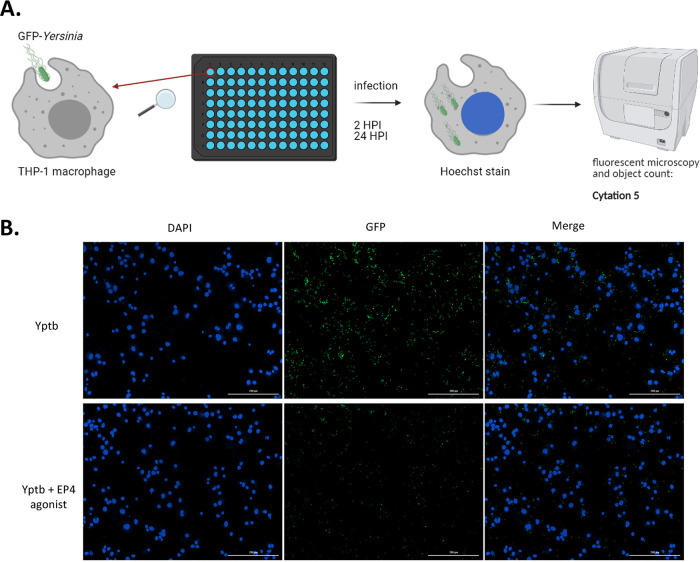
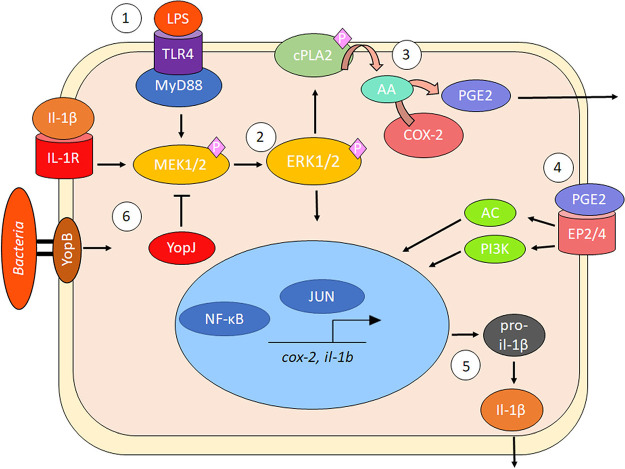
Prostaglandin E2 (PGE2) is an essential immunomodulatory lipid released by cells in response to infection with many bacteria, yet its function in macrophage-mediated bacterial clearance is poorly understood. Yersinia overall inhibits the inflammatory circuit, but its effect on PGE2 production is unknown. We hypothesized that one of the Yersinia effector proteins is responsible for the inhibition of PGE2 biosynthesis. We identified that yopB-deficient Y. enterocolitica and Y. pseudotuberculosis deficient in the secretion of virulence proteins via a type 3 secretion system (T3SS) failed to inhibit PGE2 biosynthesis in macrophages. Consistently, COX-2-mediated PGE2 biosynthesis is upregulated in cells treated with heat-killed or T3SS-deficient Y. pseudotuberculosis but diminished in the presence of a MAPK/ERK inhibitor. Mutants expressing catalytically inactive YopJ induce similar levels of PGE2 as heat-killed or ΔyopB Y. pseudotuberculosis, reversed by YopJ complementation. Shotgun proteomics discovered host pathways regulated in a YopJ-mediated manner, including pathways regulating PGE2 synthesis and oxidative phosphorylation. Consequently, this study identified that YopJ-mediated inhibition of MAPK signal transduction serves as a mechanism targeting PGE2, an alternative means of inflammasome inhibition by Yersinia. Finally, we showed that EP4 signaling supports macrophage function in clearing intracellular bacteria. In summary, our unique contribution was to determine a bacterial virulence factor that targets COX-2 transcription, thereby enhancing the intracellular survival of yersiniae. Future studies should investigate whether PGE2 or its stable synthetic derivatives could serve as a potential therapeutic molecule to improve the outcomes of specific bacterial infections. Since other pathogens encode YopJ homologs, this mechanism is expected to be present in other infections. IMPORTANCE PGE2 is a critical immunomodulatory lipid, but its role in bacterial infection and pathogen clearance is poorly understood. We previously demonstrated that PGE2 leads to macrophage polarization toward the M1 phenotype and stimulates inflammasome activation in infected macrophages. Finally, we also discovered that PGE2 improved the clearance of Y. enterocolitica. The fact that Y. enterocolitica hampers PGE2 secretion in a type 3 secretion system (T3SS)-dependent manner and because PGE2 appears to assist macrophage in the clearance of this bacterium indicates that targeting of the eicosanoid pathway by Yersinia might be an adaption used to counteract host defenses. Our study identified a mechanism used by Yersinia that obstructs PGE2 biosynthesis in human macrophages. We showed that Y. pseudotuberculosis interferes with PGE2 biosynthesis by using one of its T3SS effectors, YopJ. Specifically, YopJ targets the host COX-2 enzyme responsible for PGE2 biosynthesis, which happens in a MAPK/ER-dependent manner. Moreover, in a shotgun proteomics study, we also discovered other pathways that catalytically active YopJ targets in the infected macrophages. YopJ was revealed to play a role in limiting host LPS responses, including repression of EGR1 and JUN proteins, which control transcriptional activation of proinflammatory cytokine production such as interleukin-1β. Since YopJ has homologs in other bacterial species, there are likely other pathogens that target and inhibit PGE2 biosynthesis. In summary, our study's unique contribution was to determine a bacterial virulence factor that targets COX-2 transcription. Future studies should investigate whether PGE2 or its stable synthetic derivatives could serve as a potential therapeutic target.
Keywords: Gram-negative infection; Yersinia; eicosanoids; prostaglandin; proteomics.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
