

BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes
- PMID: 34320186
- PMCID: PMC8476166
- DOI: 10.1093/molbev/msab199
BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes
Abstract
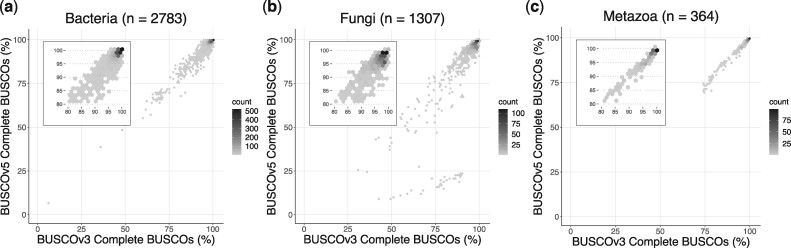
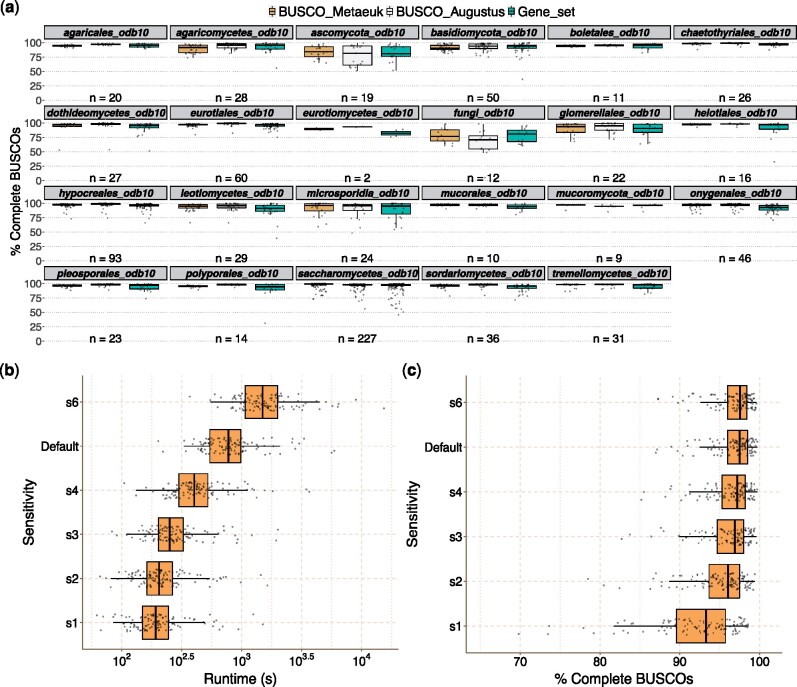
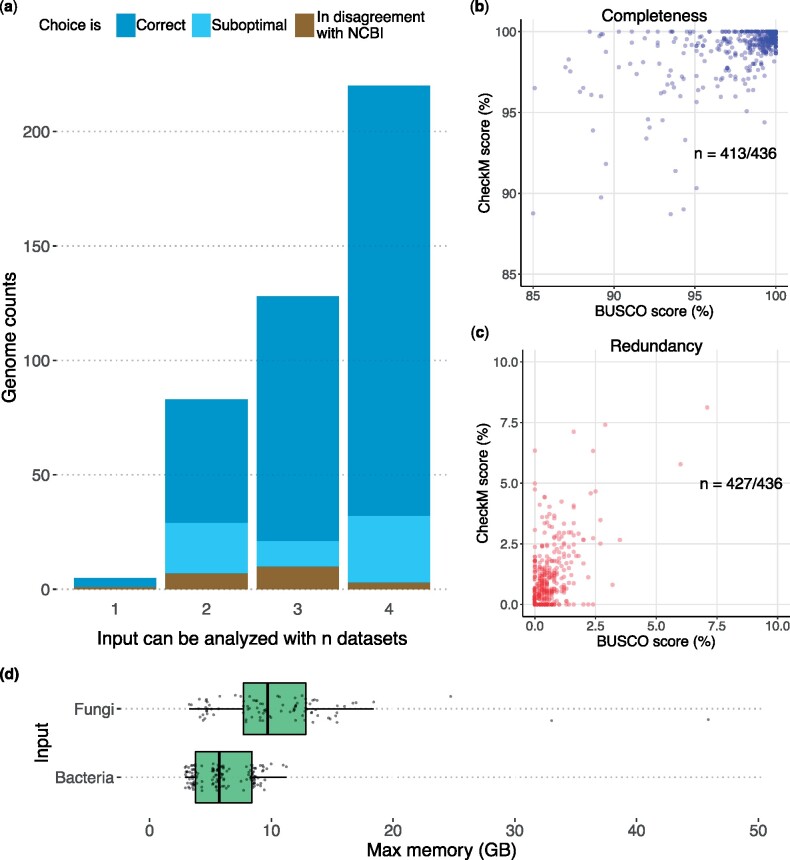
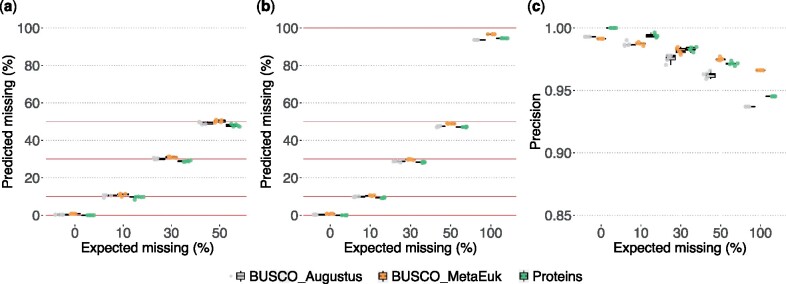
Methods for evaluating the quality of genomic and metagenomic data are essential to aid genome assembly procedures and to correctly interpret the results of subsequent analyses. BUSCO estimates the completeness and redundancy of processed genomic data based on universal single-copy orthologs. Here, we present new functionalities and major improvements of the BUSCO software, as well as the renewal and expansion of the underlying data sets in sync with the OrthoDB v10 release. Among the major novelties, BUSCO now enables phylogenetic placement of the input sequence to automatically select the most appropriate BUSCO data set for the assessment, allowing the analysis of metagenome-assembled genomes of unknown origin. A newly introduced genome workflow increases the efficiency and runtimes especially on large eukaryotic genomes. BUSCO is the only tool capable of assessing both eukaryotic and prokaryotic species, and can be applied to various data types, from genome assemblies and metagenomic bins, to transcriptomes and gene sets.
Keywords: completeness; eukaryotes; genome; metagenomes; microbes; prokaryotes; quality assessment; transcriptome; viruses.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
