

RCA2: a scalable supervised clustering algorithm that reduces batch effects in scRNA-seq data
- PMID: 34320202
- PMCID: PMC8344557
- DOI: 10.1093/nar/gkab632
RCA2: a scalable supervised clustering algorithm that reduces batch effects in scRNA-seq data
Abstract
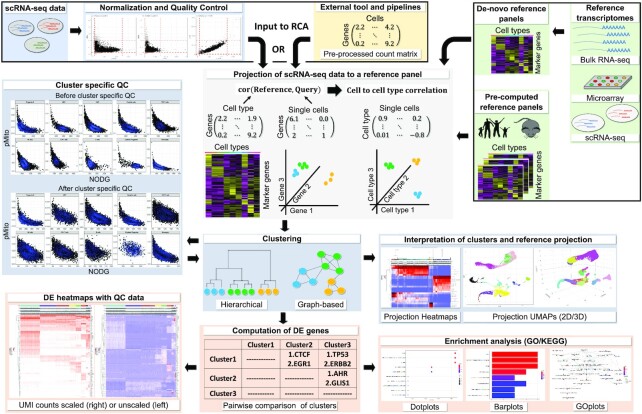
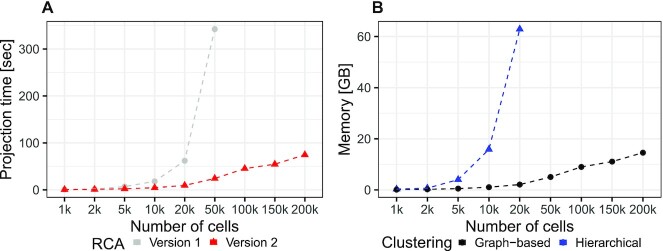
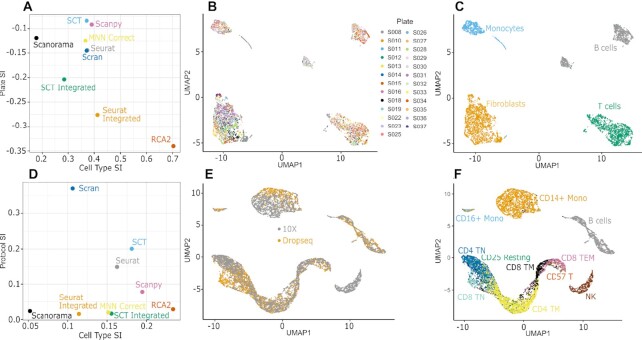
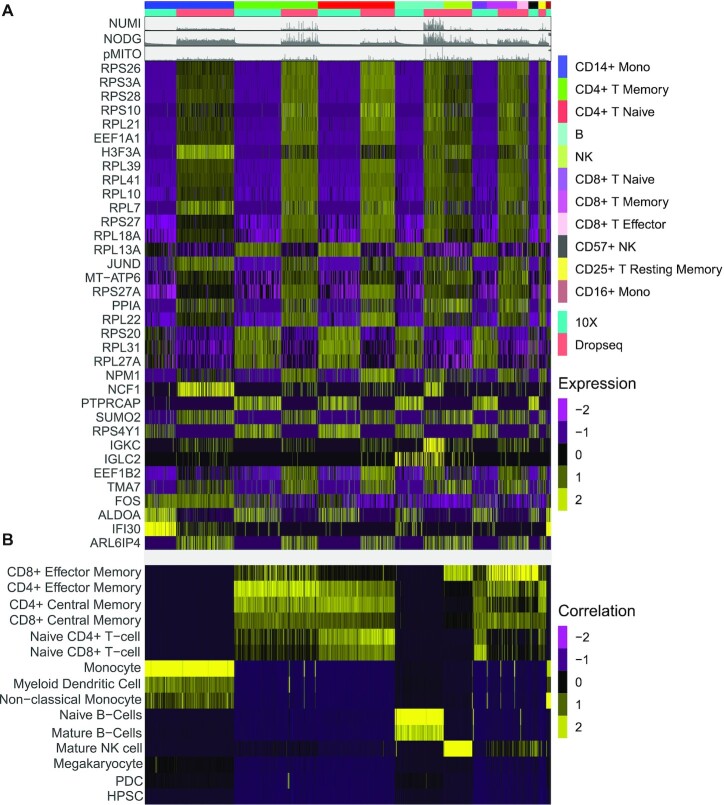
The transcriptomic diversity of cell types in the human body can be analysed in unprecedented detail using single cell (SC) technologies. Unsupervised clustering of SC transcriptomes, which is the default technique for defining cell types, is prone to group cells by technical, rather than biological, variation. Compared to de-novo (unsupervised) clustering, we demonstrate using multiple benchmarks that supervised clustering, which uses reference transcriptomes as a guide, is robust to batch effects and data quality artifacts. Here, we present RCA2, the first algorithm to combine reference projection (batch effect robustness) with graph-based clustering (scalability). In addition, RCA2 provides a user-friendly framework incorporating multiple commonly used downstream analysis modules. RCA2 also provides new reference panels for human and mouse and supports generation of custom panels. Furthermore, RCA2 facilitates cell type-specific QC, which is essential for accurate clustering of data from heterogeneous tissues. We demonstrate the advantages of RCA2 on SC data from human bone marrow, healthy PBMCs and PBMCs from COVID-19 patients. Scalable supervised clustering methods such as RCA2 will facilitate unified analysis of cohort-scale SC datasets.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
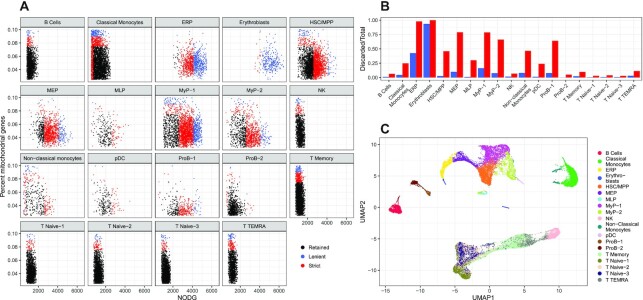
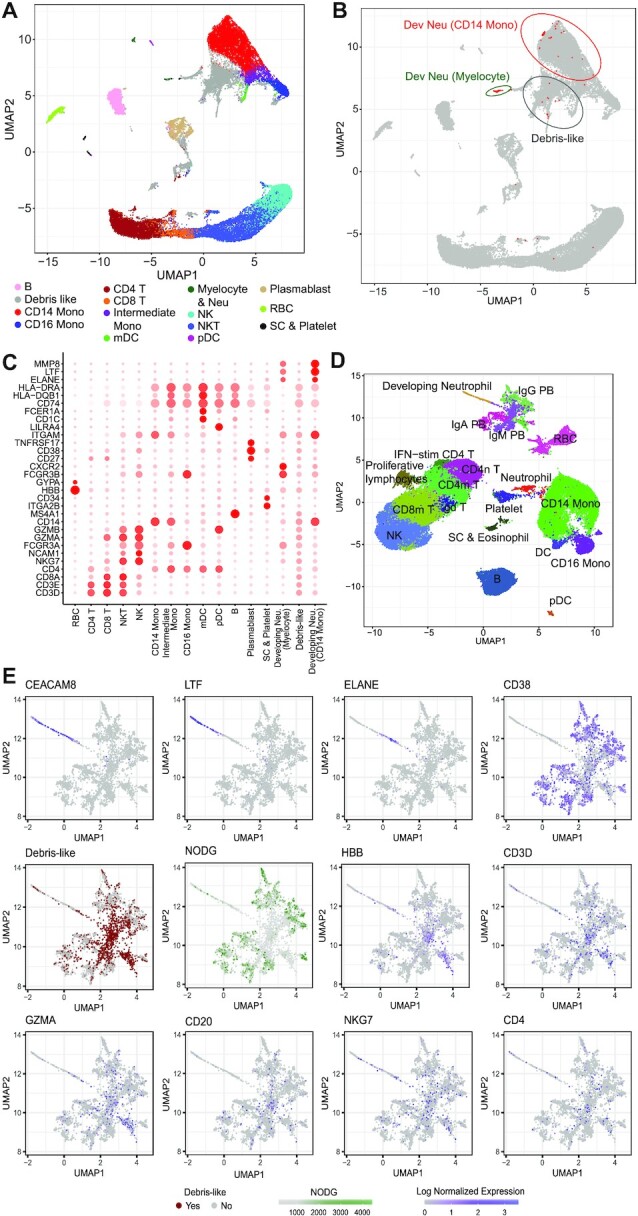
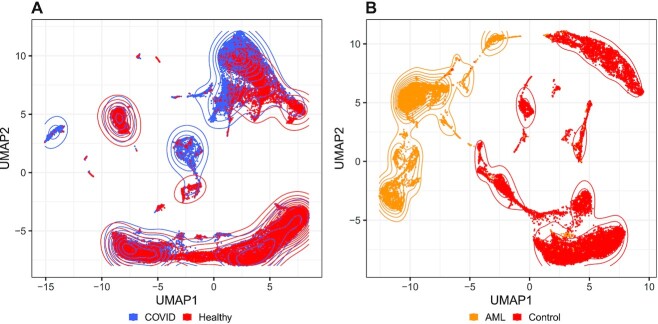
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
