

Methylation of dual-specificity phosphatase 4 controls cell differentiation
- PMID: 34320342
- PMCID: PMC9110119
- DOI: 10.1016/j.celrep.2021.109421
Methylation of dual-specificity phosphatase 4 controls cell differentiation
Abstract
Mitogen-activated protein kinases (MAPKs) are inactivated by dual-specificity phosphatases (DUSPs), the activities of which are tightly regulated during cell differentiation. Using knockdown screening and single-cell transcriptional analysis, we demonstrate that DUSP4 is the phosphatase that specifically inactivates p38 kinase to promote megakaryocyte (Mk) differentiation. Mechanistically, PRMT1-mediated methylation of DUSP4 triggers its ubiquitinylation by an E3 ligase HUWE1. Interestingly, the mechanistic axis of the DUSP4 degradation and p38 activation is also associated with a transcriptional signature of immune activation in Mk cells. In the context of thrombocytopenia observed in myelodysplastic syndrome (MDS), we demonstrate that high levels of p38 MAPK and PRMT1 are associated with low platelet counts and adverse prognosis, while pharmacological inhibition of p38 MAPK or PRMT1 stimulates megakaryopoiesis. These findings provide mechanistic insights into the role of the PRMT1-DUSP4-p38 axis on Mk differentiation and present a strategy for treatment of thrombocytopenia associated with MDS.
Trial registration: ClinicalTrials.gov NCT01496495.
Keywords: DUSP4; HUWE1; MDS; PRMT1; leukemia; megakaryocyte; myelodysplasia syndrome; p38; platlet; trombocytopenia.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.L. has served on the Scientific Advisory Board for Epi One. A.V. has received research funding from GlaxoSmithKline, Incyte, MedPacto, Novartis, Curis, and Eli Lilly and Company; has received compensation as a scientific advisor to Novartis, Stelexis Therapeutics, Acceleron Pharma, and Celgene; and has equity ownership in Stelexis Therapeutics. The remaining authors declare no competing interests. Array BioPharma provided the p38 inhibitor pexmetinib (ARRY614) and participated in its phase I study (ClinicalTrials.gov: NCT01496495).
Figures
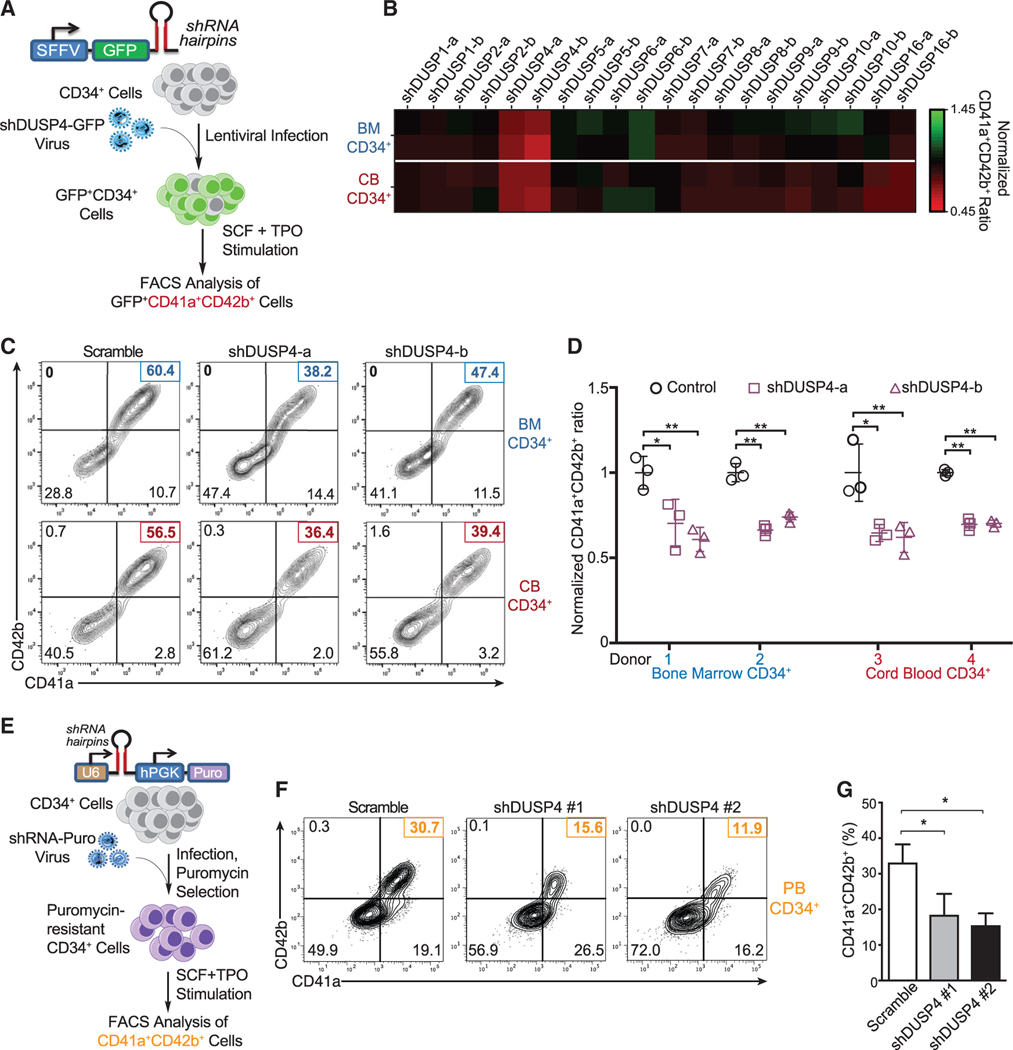
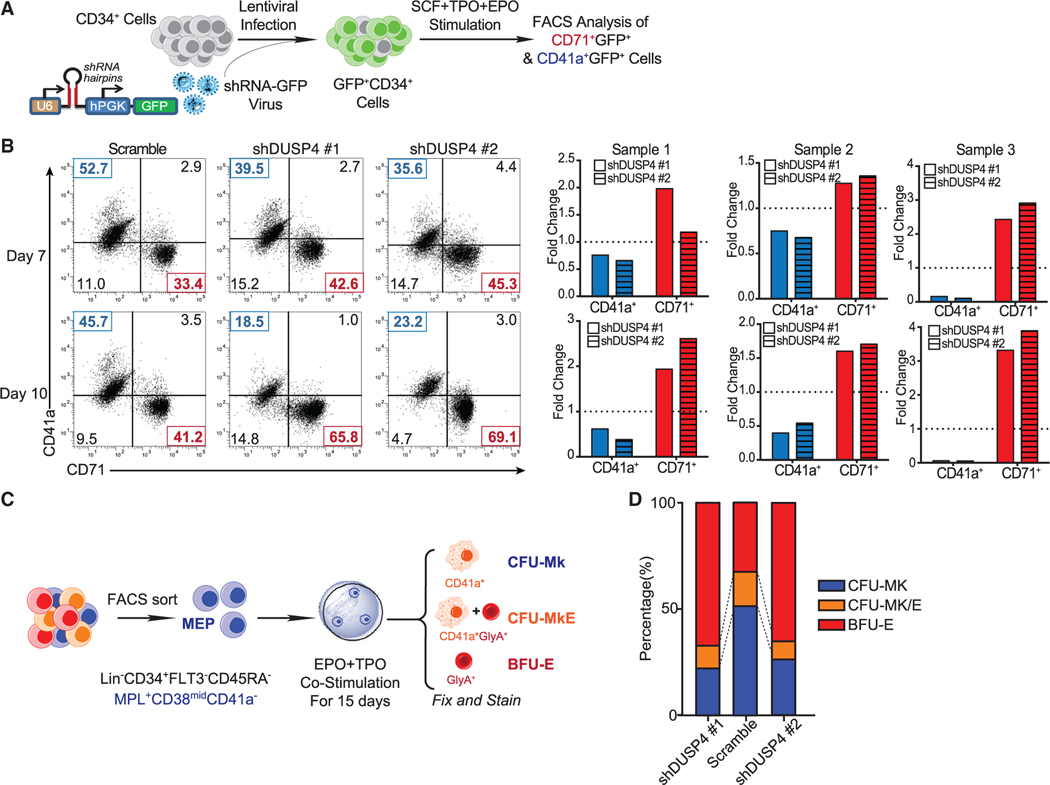
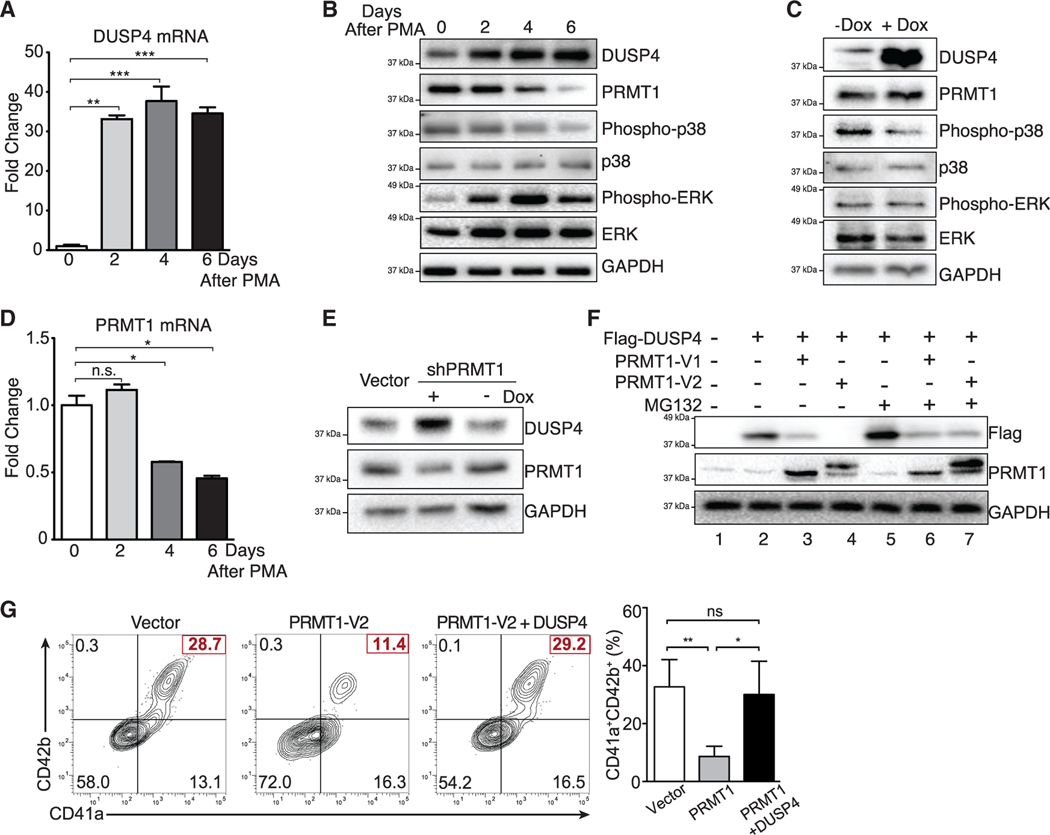
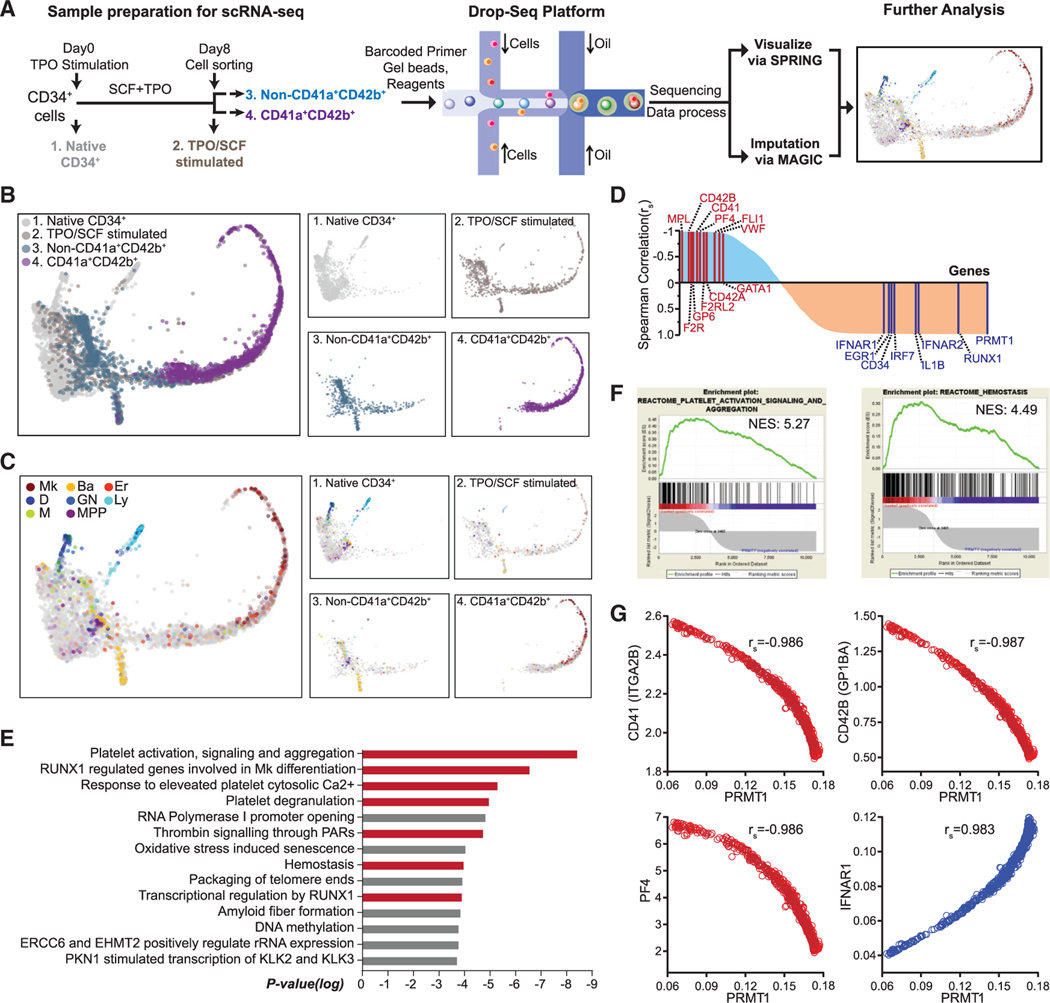
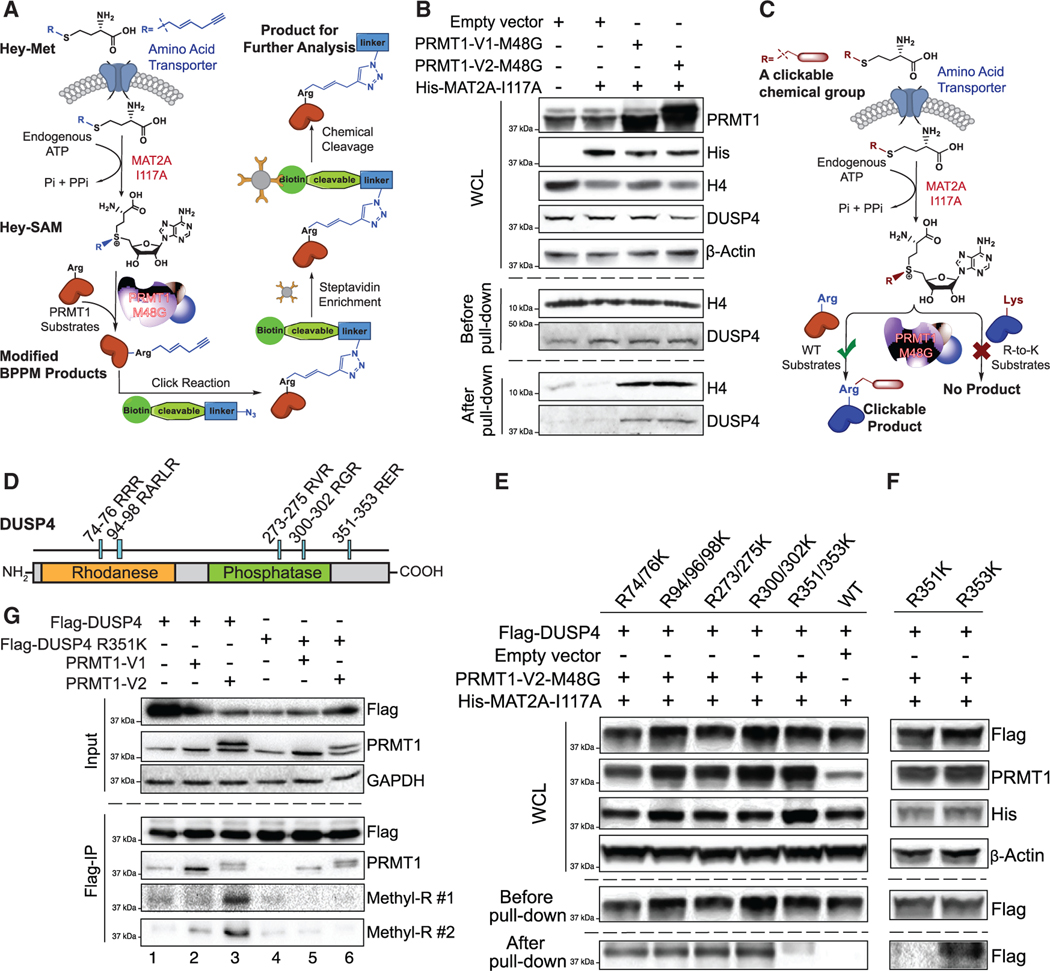
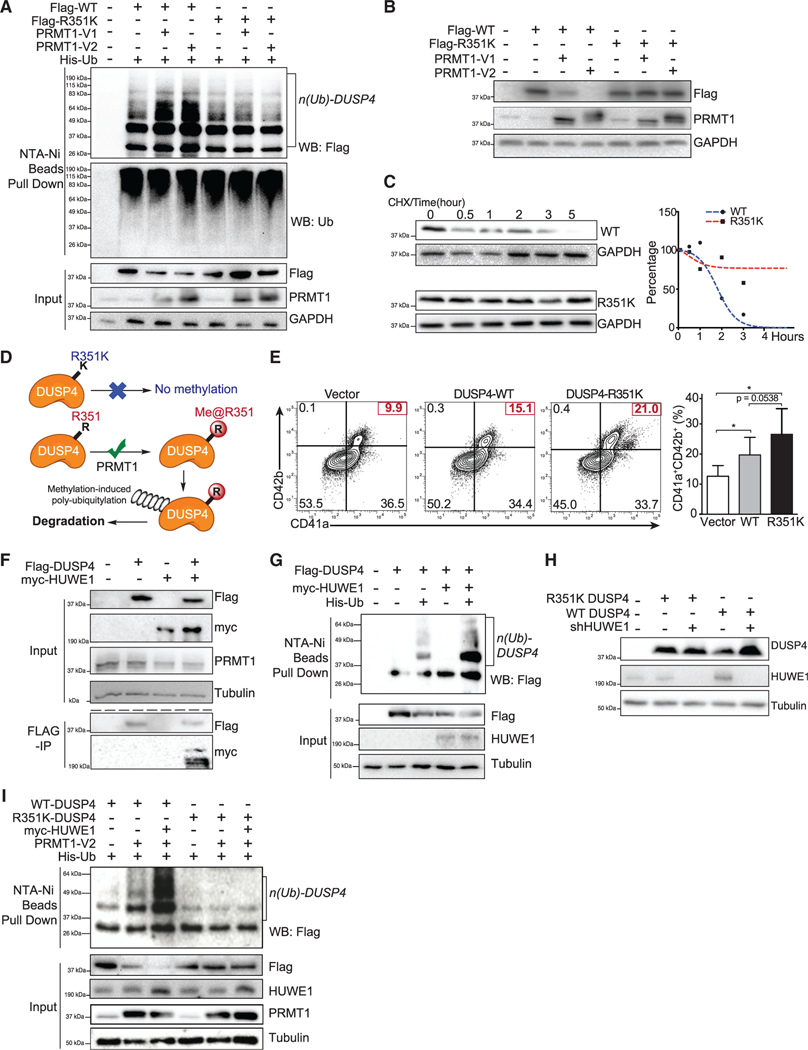
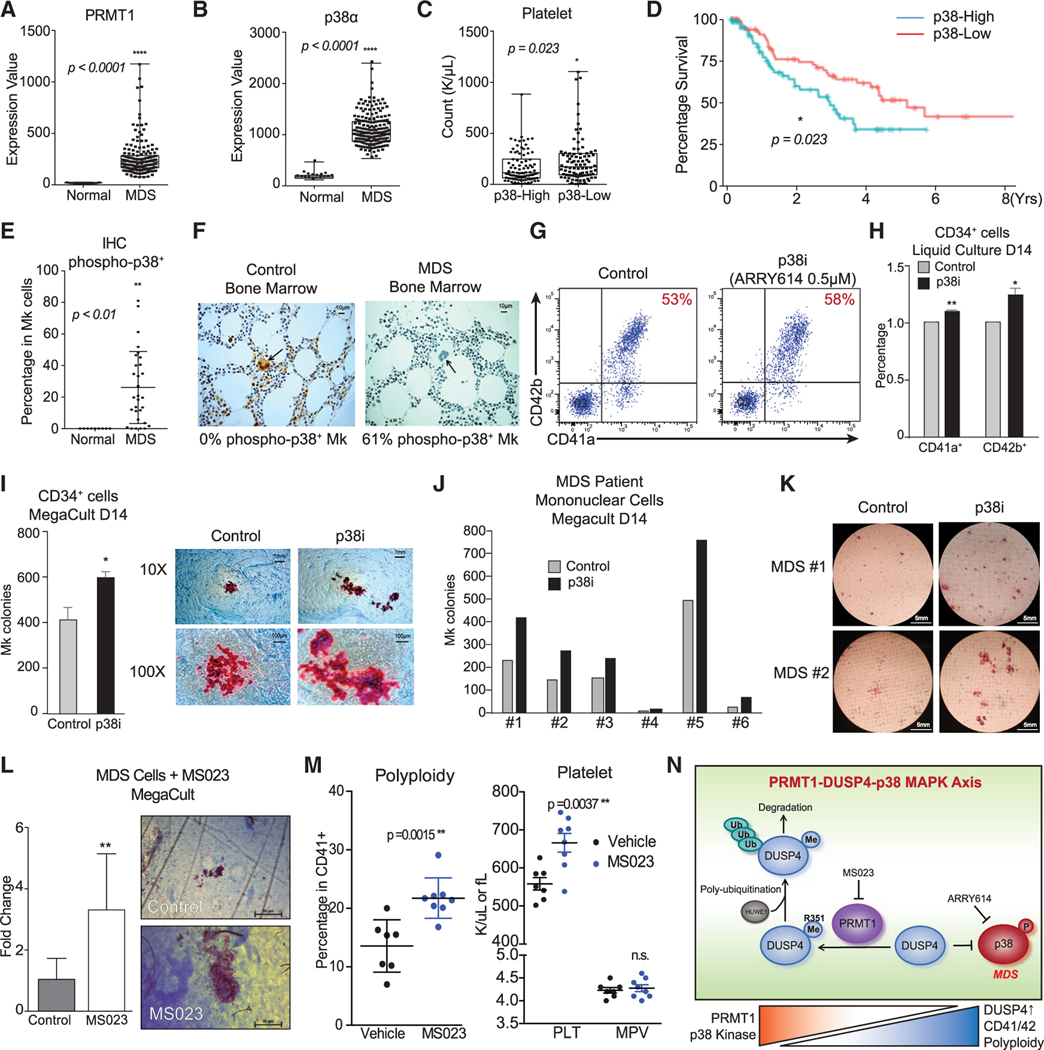
References
-
- Bachegowda L, Morrone K, Winski SL, Mantzaris I, Bartenstein M, Ramachandra N, Giricz O, Sukrithan V, Nwankwo G, Shahnaz S, et al. (2016). Pexmetinib: A novel dual inhibitor of Tie2 and p38 MAPK with efficacy in preclinical models of myelodysplastic syndromes and acute myeloid leuke mia. Cancer Res. 76, 4841–4849. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
