

O'Donnell-Luria-Rodan syndrome: description of a second multinational cohort and refinement of the phenotypic spectrum
- PMID: 34321323
- PMCID: PMC10256139
- DOI: 10.1136/jmedgenet-2020-107470
O'Donnell-Luria-Rodan syndrome: description of a second multinational cohort and refinement of the phenotypic spectrum
Abstract
Background: O'Donnell-Luria-Rodan syndrome (ODLURO) is an autosomal-dominant neurodevelopmental disorder caused by pathogenic, mostly truncating variants in KMT2E. It was first described by O'Donnell-Luria et al in 2019 in a cohort of 38 patients. Clinical features encompass macrocephaly, mild intellectual disability (ID), autism spectrum disorder (ASD) susceptibility and seizure susceptibility.
Methods: Affected individuals were ascertained at paediatric and genetic centres in various countries by diagnostic chromosome microarray or exome/genome sequencing. Patients were collected into a case cohort and were systematically phenotyped where possible.
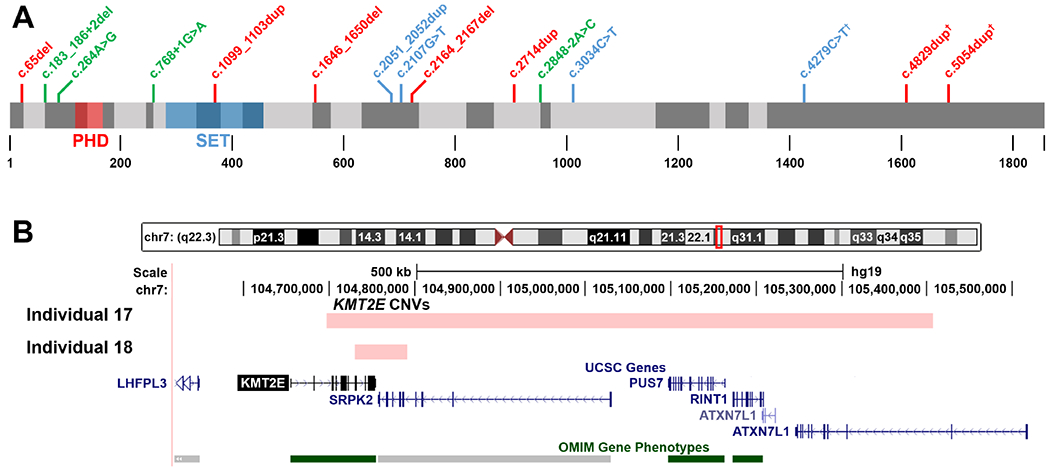
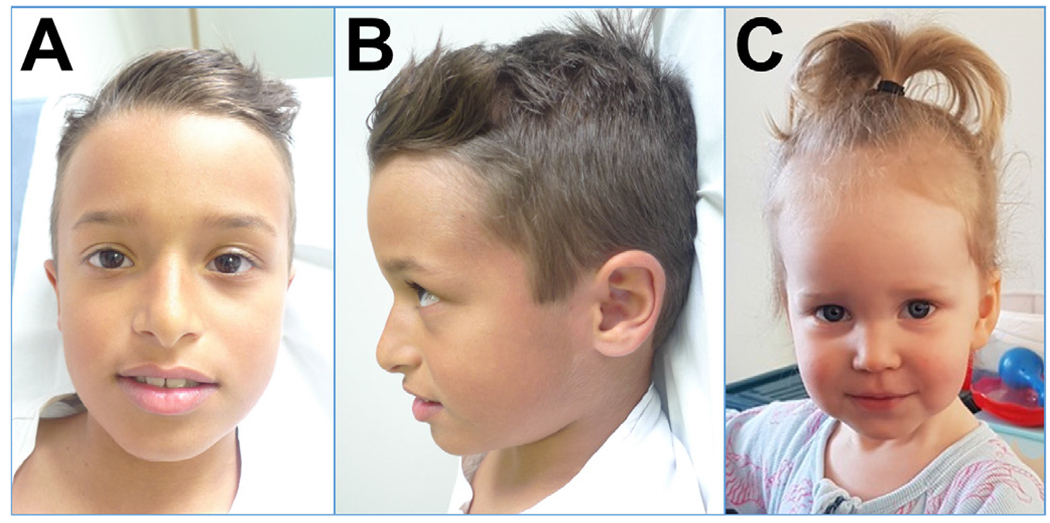
Results: We report 18 additional patients from 17 families with genetically confirmed ODLURO. We identified 15 different heterozygous likely pathogenic or pathogenic sequence variants (14 novel) and two partial microdeletions of KMT2E. We confirm and refine the phenotypic spectrum of the KMT2E-related neurodevelopmental disorder, especially concerning cognitive development, with rather mild ID and macrocephaly with subtle facial features in most patients. We observe a high prevalence of ASD in our cohort (41%), while seizures are present in only two patients. We extend the phenotypic spectrum by sleep disturbances.
Conclusion: Our study, bringing the total of known patients with ODLURO to more than 60 within 2 years of the first publication, suggests an unexpectedly high relative frequency of this syndrome worldwide. It seems likely that ODLURO, although just recently described, is among the more common single-gene aetiologies of neurodevelopmental delay and ASD. We present the second systematic case series of patients with ODLURO, further refining the mutational and phenotypic spectrum of this not-so-rare syndrome.
Keywords: behavioural; genetic counselling; genetics; human genetics; mutation.
© Author(s) (or their employer(s)) 2022. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y-H, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron 2012;74:285–99. - PMC - PubMed
-
- Wang T, Guo H, Xiong B, Stessman HAF, Wu H, Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, Vives L, Xia L, Tang M, Ou J, Chen B, Shen Y, Xun G, Long M, Lin J, Kronenberg ZN, Peng Y, Bai T, Li H, Ke X, Hu Z, Zhao J, Zou X, Xia K, Eichler EE. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 2016;7:13316. - PMC - PubMed
-
- Guo H, Wang T, Wu H, Long M, Coe BP, Li H, Xun G, Ou J, Chen B, Duan G, Bai T, Zhao N, Shen Y, Li Y, Wang Y, Zhang Y, Baker C, Liu Y, Pang N, Huang L, Han L, Jia X, Liu C, Ni H, Yang X, Xia L, Chen J, Shen L, Li Y, Zhao R, Zhao W, Peng J, Pan Q, Long Z, Su W, Tan J, Du X, Ke X, Yao M, Hu Z, Zou X, Zhao J, Bernier RA, Eichler EE, Xia K. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol Autism 2018;9:64. - PMC - PubMed
-
- Dong S, Walker MF, Carriero NJ, DiCola M, Willsey AJ, Ye AY, Waqar Z, Gonzalez LE, Overton JD, Frahm S, Keaney JF, Teran NA, Dea J, Mandell JD, Hus Bal V, Sullivan CA, DiLullo NM, Khalil RO, Gockley J, Yuksel Z, Sertel SM, Ercan-Sencicek AG, Gupta AR, Mane SM, Sheldon M, Brooks AI, Roeder K, Devlin B, State MW, Wei L, Sanders SJ. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep 2014;9:16–23. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases