

An erythroid-to-myeloid cell fate conversion is elicited by LSD1 inactivation
- PMID: 34324630
- PMCID: PMC8569417
- DOI: 10.1182/blood.2021011682
An erythroid-to-myeloid cell fate conversion is elicited by LSD1 inactivation
Abstract
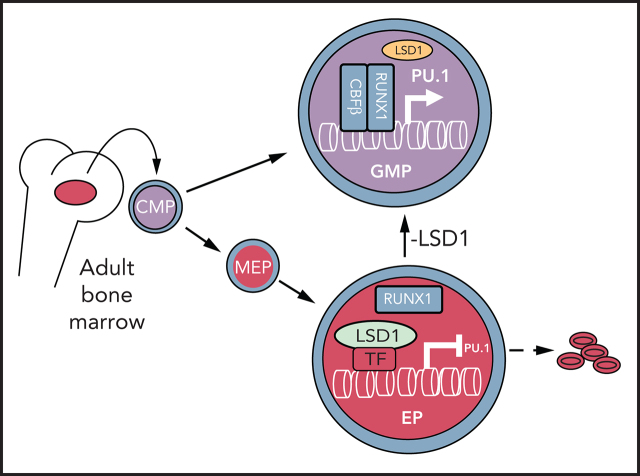
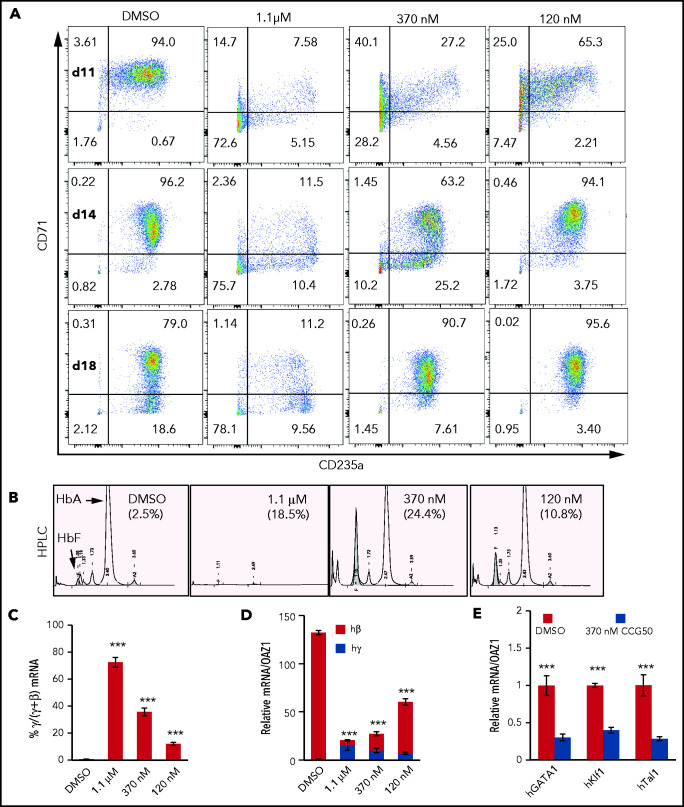
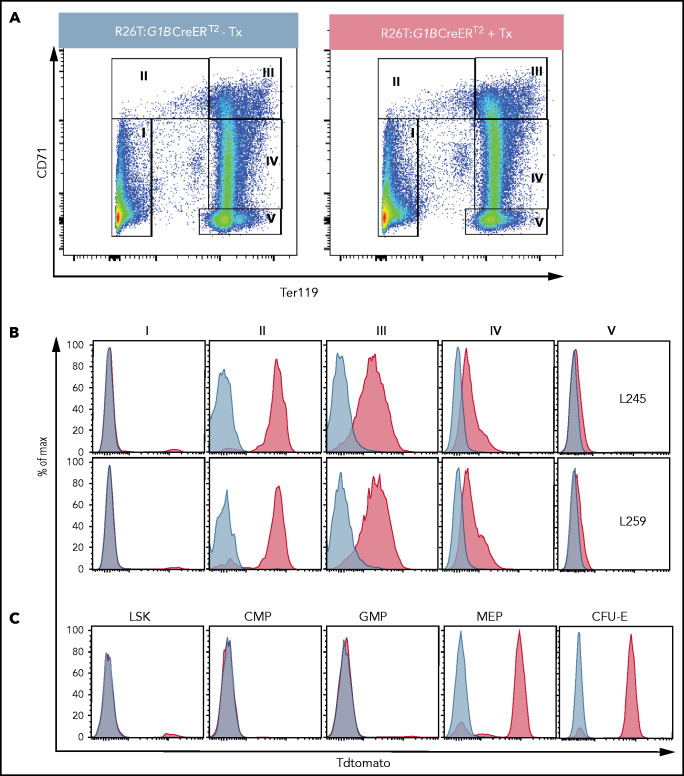
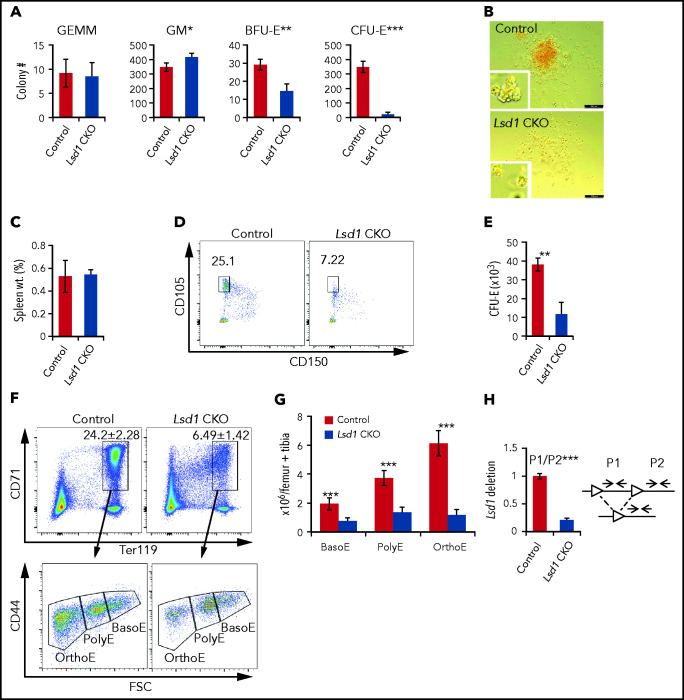
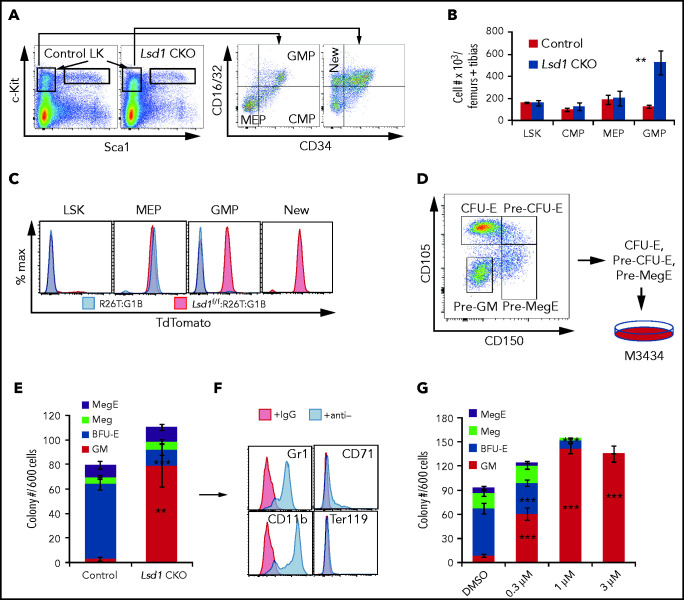
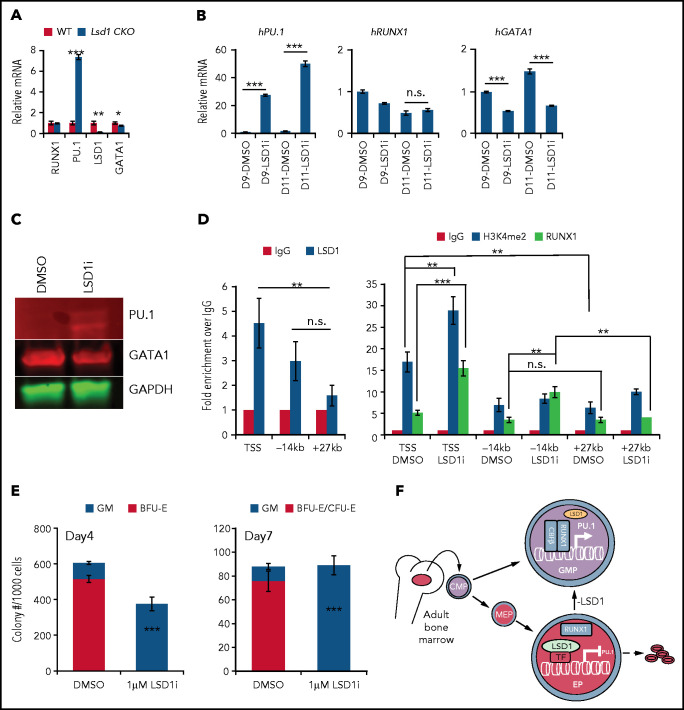
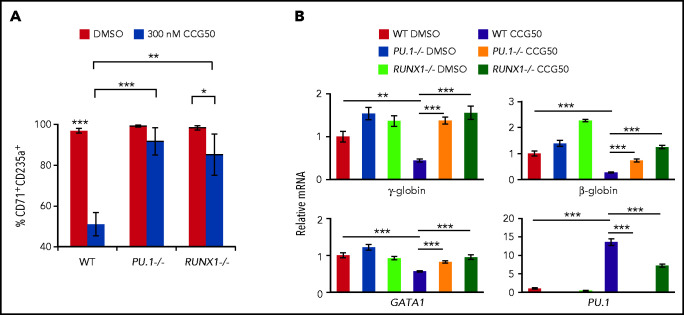
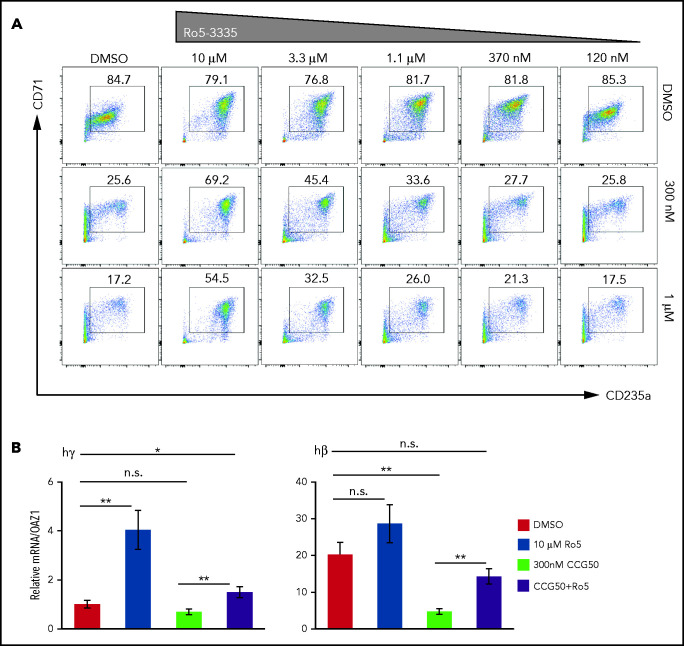
Histone H3 lysine 4 methylation (H3K4Me) is most often associated with chromatin activation, and removing H3K4 methyl groups has been shown to be coincident with gene repression. H3K4Me demethylase KDM1a/LSD1 is a therapeutic target for multiple diseases, including for the potential treatment of β-globinopathies (sickle cell disease and β-thalassemia), because it is a component of γ-globin repressor complexes, and LSD1 inactivation leads to robust induction of the fetal globin genes. The effects of LSD1 inhibition in definitive erythropoiesis are not well characterized, so we examined the consequences of conditional inactivation of Lsd1 in adult red blood cells using a new Gata1creERT2 bacterial artificial chromosome transgene. Erythroid-specific loss of Lsd1 activity in mice led to a block in erythroid progenitor differentiation and to the expansion of granulocyte-monocyte progenitor-like cells, converting hematopoietic differentiation potential from an erythroid fate to a myeloid fate. The analogous phenotype was also observed in human hematopoietic stem and progenitor cells, coincident with the induction of myeloid transcription factors (eg, PU.1 and CEBPα). Finally, blocking the activity of the transcription factor PU.1 or RUNX1 at the same time as LSD1 inhibition rescued myeloid lineage conversion to an erythroid phenotype. These data show that LSD1 promotes erythropoiesis by repressing myeloid cell fate in adult erythroid progenitors and that inhibition of the myeloid-differentiation pathway reverses the lineage switch induced by LSD1 inactivation.
© 2021 by The American Society of Hematology.
Figures
Comment in
-
Epigenetic plasticity of erythroid progenitors.Blood. 2021 Nov 4;138(18):1646-1648. doi: 10.1182/blood.2021013087. Blood. 2021. PMID: 34735002 Free PMC article. No abstract available.
References
-
- Murayama M. Molecular mechanism of red cell “sickling”. Science. 1966;153(3732): 145-149. - PubMed
-
- Platt OS, Brambilla DJ, Rosse WF, et al. . Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644. - PubMed
-
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619. - PubMed
-
- Dedoussis GV, Sinopoulou K, Gyparaki M, Loutradis A.. Fetal hemoglobin expression in the compound heterozygous state for −117 (G→A) Agamma HPFH and IVSII-745 (C→G) beta+ thalassemia: a case study. Am J Hematol. 1999;61(2):139-143. - PubMed
-
- Dedoussis GV, Sinopoulou K, Gyparaki M, Loutradis A.. Fetal hemoglobin expression in the compound heterozygous state for −117 (G→A) Agamma HPFH and IVS-1 nt 110 (G→A) beta+ thalassemia: a case study. Eur J Haematol. 2000;65(2):93-96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
