

Genetics of Cardiomyopathy: Clinical and Mechanistic Implications for Heart Failure
- PMID: 34327881
- PMCID: PMC8484993
- DOI: 10.4070/kcj.2021.0154
Genetics of Cardiomyopathy: Clinical and Mechanistic Implications for Heart Failure
Abstract
Genetics has played an important role in the understanding of different cardiomyopathies, and the field of heart failure (HF) genetics is progressing rapidly. Much research has also focused on distinguishing markers of risk in patients with cardiomyopathy using genetic testing. While these efforts currently remain incomplete, new genomic technologies and analytical strategies provide promising opportunities to further explore the genetic architecture of cardiomyopathies, afford insight into the early manifestations of cardiomyopathy, and help define the molecular pathophysiological basis for cardiac remodeling. Cardiovascular physicians should be fully aware of the utility and potential pitfalls of incorporating genetic test results into pre-emptive treatment strategies for patients in the preliminary stages of HF. Future work will need to be directed towards elucidating the biological mechanisms of both rare and common gene variants and environmental determinants of plasticity in the genotype-phenotype relationship. This future research should aim to further our ability to identify, diagnose, and treat disorders that cause HF and sudden cardiac death in young patients, as well as prioritize improving our ability to stratify the risk for these patients prior to the onset of the more severe consequences of their disease.
Keywords: Cardiomyopathy; Dilated cardiomyopathy; Genetics; Heart failure; Hypertrophic cardiomyopathy.
Copyright © 2021. The Korean Society of Cardiology.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
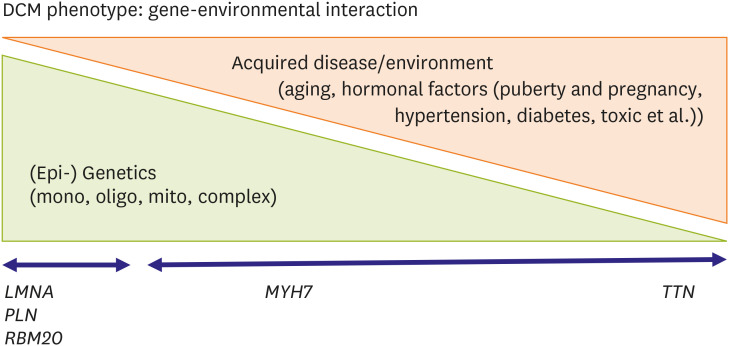
References
-
- Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: a translational review of current literature. J Intern Med. 2019;286:362–372. - PubMed
-
- Sabater-Molina M, Pérez-Sánchez I, Hernández Del Rincón JP, Gimeno JR. Genetics of hypertrophic cardiomyopathy: a review of current state. Clin Genet. 2018;93:3–14. - PubMed
-
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
